GP1BA 基因复合杂合变异致Bernard-Soulier 综合征1 例报告
2022-01-20沈笛颖张晶樱余金丹
王 燕 沈笛颖 张晶樱 余金丹
浙江大学医学院附属儿童医院 国家儿童健康与疾病临床医学研究中心 国家儿童区域医疗中心(浙江杭州 310000 )
Bernard-Soulier 综合征(Bernard-Soulier syndrome,BSS)是一种极为罕见的遗传性凝血疾病,其特征是巨大血小板、血小板减少和出血时间延长;BSS 患病率约为 1/100 万。BSS 的临床表现包括早期的皮肤黏膜出血,牙龈出血,鼻出血及月经量增多等,颅内出血及消化道出血少见[1]。由于BSS 较为罕见且缺乏特异性表型,因此早期诊断较困难,平均诊断年龄为16 岁。由于 BSS 与免疫性血小板减少症(ITP)在新生儿期临床表现类似,许多BSS 患者在新生儿期易被诊断为ITP,予激素治疗,甚至接受脾切除术。因此,了解BSS 的表现特征,结合分子遗传学检测,对其进行早期诊断十分必要[2]。
1 临床资料
患儿,男,5 岁,因鼻出血1 天,呕血半天,于2018 年1 月就诊于浙江大学医学院附属儿童医院。患儿系G1P1,足月顺产,出生体质量3.4 kg。患儿自新生儿期就出现血小板计数减少,多次予静脉滴注丙种球蛋白治疗,血小板维持在(25~40)×109/L。一年余前曾予泼尼松片治疗,效果不佳,已停止治疗1 年。患儿父母无血小板减少病史,非近亲结婚。入院体格检查:体温37.5 ℃,心率136 次/min,呼吸28次/min,血压124/71 mmHg。四肢可见数颗陈旧性皮疹,略高出皮面,压之不褪色;咽充血,咽腭弓处可见暗红色瘀斑;腹软,肝脾肋下未触及。实验室检查:血常规示血小板计数28×109/L,平均血小板体积12.1 fL;超敏C 反应蛋白11.0 mg/L;凝血酶原时间11.9 s,活化部分凝血活酶时间<17.0 s,纤维蛋白原1.84 g/L,凝血酶时间21.9 s;遗传代谢病筛查未见异常。入院后查血小板抗体75.36%。骨髓穿刺:巨核细胞数量中等,全片共见巨核细胞61 个,以颗粒型为主,散在及成堆血小板可见;未见其他异常细胞。流式细胞术检测示血小板膜糖蛋白Ⅰbα(platelet glycoprotein Ⅰb alpha,GP Ⅰbα)表达水平为42.4%,显著低于正常水平(图1)。

图1 GP bα流式细胞术分析
入院后予丙种球蛋白、甲基泼尼松龙静脉滴注治疗,患儿血小板上升不明显。为进一步明确诊断,经医学伦理审核并获得患儿家长知情同意后,取患儿及父母外周血2 mL,提取DNA后对患儿DNA进行全外显子基因检测,并将其父母DNA 进行家系验证。结果显示,患儿GP 1 BA基因外显子区域存在2 处杂合变异。一处为987 位的核苷酸由鸟嘌呤突变为腺嘌呤A(c.987 G>A),导致329 位的色氨酸突变为终止密码子,导致氨基酸编码提前终止,该位点已有文献报道为致病性变异,经美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)指南判定,该位点为致病性变异;另一处为523 到525 位的核苷酸缺失(c.523_525delAAC),导致175 位的天冬酰胺缺失,为整码变异,在多个正常人群数据库中均未见携带,该位点未见文献报道,经ACMG 指南判定,该位点为临床意义未明变异,因此该患儿被确诊为BSS。经Sanger家系验证,c.987G>A变异来源于母亲;c.523_525delAAC 变异来源于父亲,为复合杂合变异(图2),患儿父母血小板计数均在正常范围,无临床表现。诊断明确后,未再予药物治疗,随访1年,期间患儿血小板计数维持在(40~60)×109/L,有数次鼻出血,按压后能很快缓解,外伤后有皮肤黏膜出血,能自行消退,无消化道大出血、颅内出血发生。
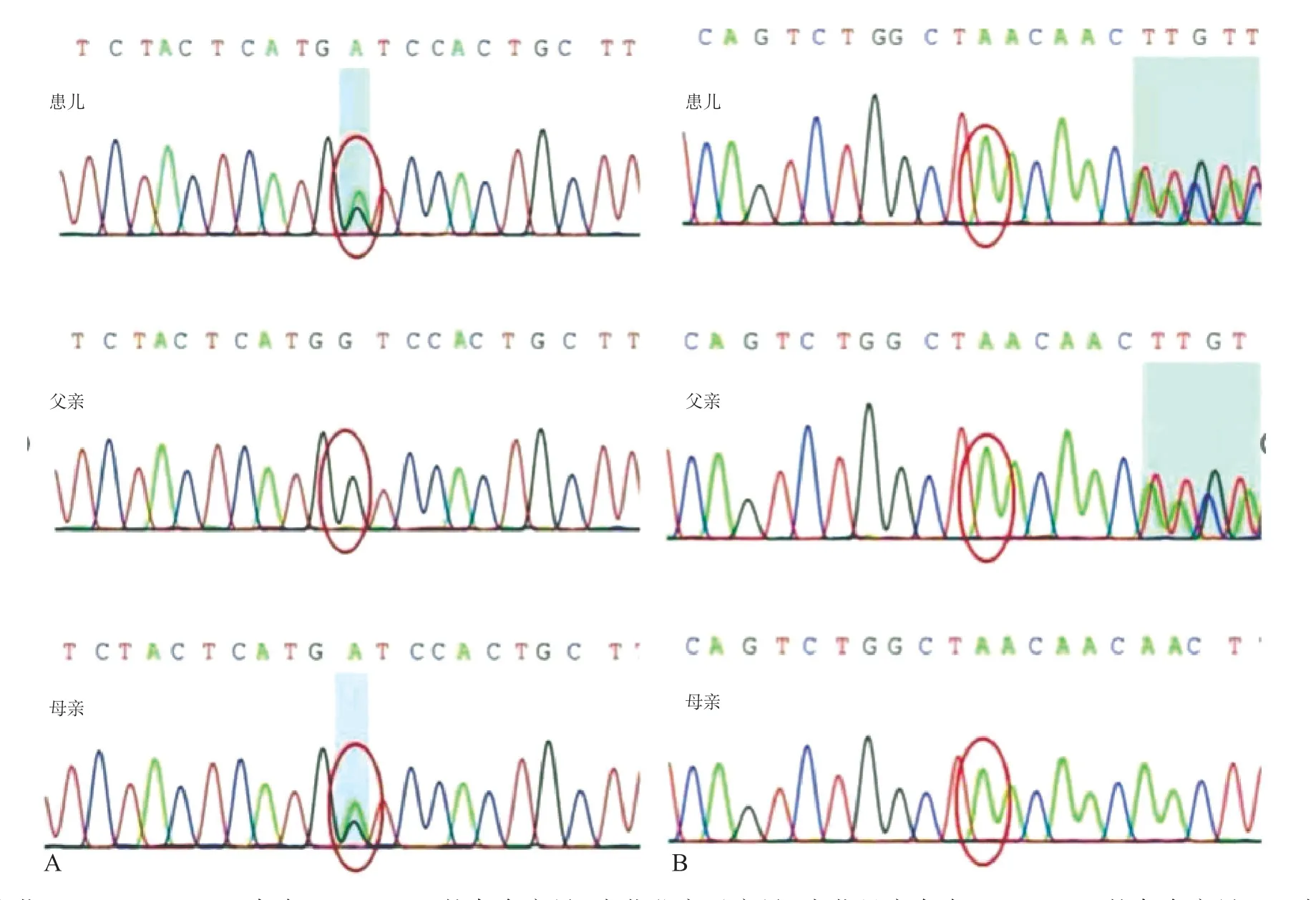
图2 患儿及父母GP bα外显子Sanger测序图
2 讨论
BSS 又名巨大血小板综合征,目前尚无明确的诊断标准,当临床上出现出血时间延长、血小板减少、血小板体积巨大、瑞斯托霉素不能诱导血小板聚集、缺乏血小板糖蛋白)等,需考虑BSS,但确诊还需依靠分子遗传学检测。
目前发现,BSS 与GP 1 BA、GP 1 BB和GP 9相关,这3个基因分别编码复合物组成蛋白复合物为血小板受体,可与凝血因子结合,帮助血小板黏附到损伤部位,复合物缺陷则会导致血小板黏附异常,增加出血倾向。在对211例散发BSS患者的基因检测中,92例(43.6%)检测到GP9变异,60例(28.4%)检测到GP1BA变异,59例(28.0%)检测到GP1BB变异[2],表明GP1BA、GP1BB以及GP9变异较为常见,尚未见GPV变异导致BBS的报道。
GP1BA基因位于17p13.2,全长2.8 kb,由652个氨基酸构成,包含2个外显子。GP1BA包含3个结构域,包括N-cap-LRR-C-cap结构域,跨膜结构域以及胞质结构域。其中,N-cap-LRR-C-cap结构域可与细胞膜结合,跨膜结构域可与亚基的跨膜结构域相互作用并参与复杂的组装,胞质结构域与细胞骨架相互作用决定血小板大小[2,5]。通过与血小板表面的胶原受体GPⅥ结合,使血小板黏附在血管壁受损部位,通过与凝血酶的结合促进血小板对低浓度凝血酶的反应[6]。
本例患儿临床表现包括鼻出血、呕血,肝脾无异常,实验室检查提示血小板减少,血小板体积大,当时考虑ITP,予以止血、丙种球蛋白及激素治疗后,血小板升高但仍低于正常范围,并反复出现血小板减少伴出血,需考虑其他出凝血疾病。蛋白表达水平仅为42.4%,显著低于正常水平,应考虑BSS,并完善基因检测。本例患儿检测到GP1BA基因上c.987G>A 和c.523_525delAAC两处变异,c.987 G>A 为终止变异,使329 位的色氨酸变异为终止密码子,导致蛋白截短,该变异可导致跨膜结构域以及胞质结构域功能丧失,c.523_525delAAC 位于N-cap-LRR-C-cap结构域,因175 位的天冬酰胺缺失,可能影响N-cap-LRR-Ccap 结构域的正常功能。结合临床表现、流式细胞术及分子遗传学检测结果,本例患儿可明确诊断为BSS。
由于BSS 临床多表现为血小板减少,缺少表型特异性,因此BSS 需与其他各种出血性疾病进行鉴别诊断。常见鉴别诊断疾病包括:①ITP,本例患儿发病初期被诊断为ITP,且对静脉滴注免疫球蛋白和类固醇等一线治疗效果不佳,则需进行流式细胞仪检测膜糖蛋白水平,与ITP 鉴别诊断。避免部分患儿被误诊为ITP,导致激素药物治疗无效,甚至接受脾切除术[7]。②血管性血友病(von Willebrand’s disease,vWD),vWD临床出血表现相差很大,实验室检查提示出血时间延长与活化的部分凝血活酶时间延长,2B型vWD的血小板也增大,同时低浓度瑞斯托霉素可增强血小板聚集,往往存在不同程度的血小板减少。而BSS患儿瑞斯托霉素不能诱导血小板聚集。③MYH 9 综合征,本例患儿血小板减少伴出血,临床治疗效果不佳,还需考虑其他遗传性血小板异常疾病,MYH9综合征临床上可表现为神经性耳聋、白内障和肾炎,外周血中性粒细胞内可见Dohle包涵体,临床中需详细询问病史及家族史,开阔诊断思路。
BSS 的治疗目前尚无统一标准,针对BSS 患儿需要精心护理,避免创伤导致出血,遵循预防措施[6],如手术需提前告知,以最大程度地减少出血事件,同时需要尽量避免并发症。对于严重BSS 患儿,需经常输注血小板以控制严重创伤或手术引起的危及生命的出血,或者进行人类干细胞移植。对于轻中度出血性疾病患者,除血小板输注外,还可采用抗纤维蛋白溶解药物、去氨加压素、雌激素等治疗[8-9]。
综上,BSS病例数量较少,国内报道约10例[6],临床认识不足,容易延误诊断,基因检测有助于 BSS患儿早期诊断。
