异柠檬酸脱氢酶1(IDH1)抑制剂的三维定量构效关系和分子对接研究
2022-01-12赵学敏郭红梅全纹萱
赵学敏,郭红梅,余 娜,赵 镜,付 乐,全纹萱,舒 茂
(重庆理工大学 药学与生物工程学院,重庆 400054)
异柠檬酸脱氢酶(IDH)是细胞代谢过程中的关键酶,在很多生理过程中都发挥着重要作用,如血管生成、代谢等[1-2]。它包括3种同工酶,分别是IDH1、IDH2、IDH3[2]。IDH突变对肿瘤发展有很重要影响。IDH1在细胞生命活动过程中扮演着重要角色,是一种不可或缺的酶。2-羟基戊二酸(2-HG)是1DH1突变时的产物。由于2-HG与α-酮戊二酸结构极其相似,而累计的2-GH能够发挥抑制双加氧酶的作用,而且2-HG 本身也是一种肿瘤细胞代谢产物,容易引起细胞分化障碍[3-4],如可以抑制氨基酸去甲基化酶,进而破坏组蛋白产生去甲基化反应,抑制参与表观遗传调节及调节细胞信号等[5-7],从而促进肿瘤的发生。在近几年的报道中发现IDH1的突变存在于多种血液病及癌症中,如急性髓性白血病(AML)[8-10]、脑胶质瘤[11-12]和骨髓增生异常综合征(MDS)。IDHl可以抑制IDHl突变体,降低2-HG的产生,从而使其达到正常水平,发挥抑制肿瘤的作用[13]。小分子抑制突变IDH1(mIDH1)已被临床验证为治疗急性髓细胞性白血病和多发实体瘤的有前途的治疗方法[14-16]。因此,IDH1抑制剂可以有效地治疗癌症。
计算机辅助药物设计已有多年的历史,其中三维定量构效关系和分子对接广泛应用于先导化合物的设计和筛选[17-21]。三维定量构效关系(3D-QSAR)利用多种描述符与其生物活性搭建结构与活性的线性关系,如多元线性回归和偏最小二乘法[22-23]。Lin等[15-16]设计合成的一系列喹啉酮类衍生物能够选择性地抑制IDH1的突变,选出的化合物具有治疗多种癌症的潜力。但是目前还未有文章报道结构与活性之间的关系。本研究可为开发新的抑制剂提供理论参考,通过采用3D-QSAR和分子对接研究结构与活性之间的关系。
1 实验部分
1.1 化合物及其活性数据
研究了来自文献的38个喹啉酮类衍生物,其分子结构及活性数据都从文献中获取[15-16]。参考结构的多样性及活性的范围,将38个化合物中的30个化合物分为训练集,8个分为测试集。为方便计算,将IC50(μmol/L)转换为pIC50(pIC50-LogIC50)。利用Sybyl-X 2.0软件进行一系列的准备工作:利用Powell能量梯度法优化,优化次数最大10 000次,收敛标准为能量变化不高于0.005 kJ(mol·Å)-1,加上Gasteiger-Huckel电荷和Tripos力场,其余数值均为默认。38个喹啉酮类IDH1抑制剂的结构和活性数据参见表1。
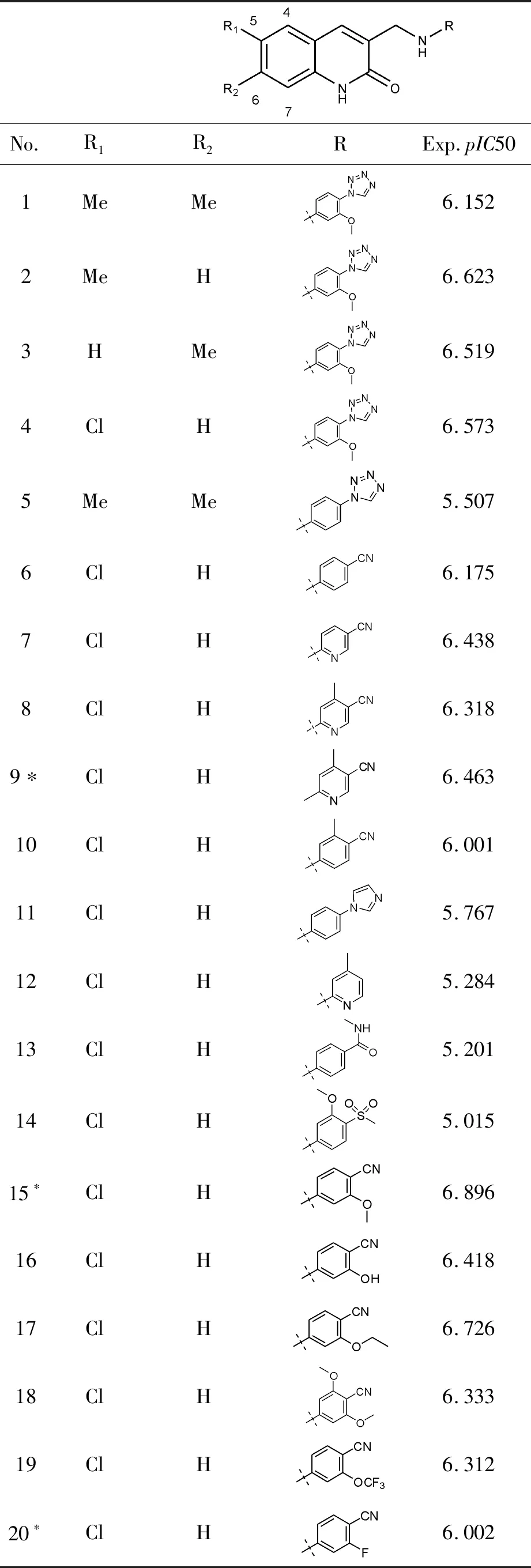
表1 化合物的活性值与结构
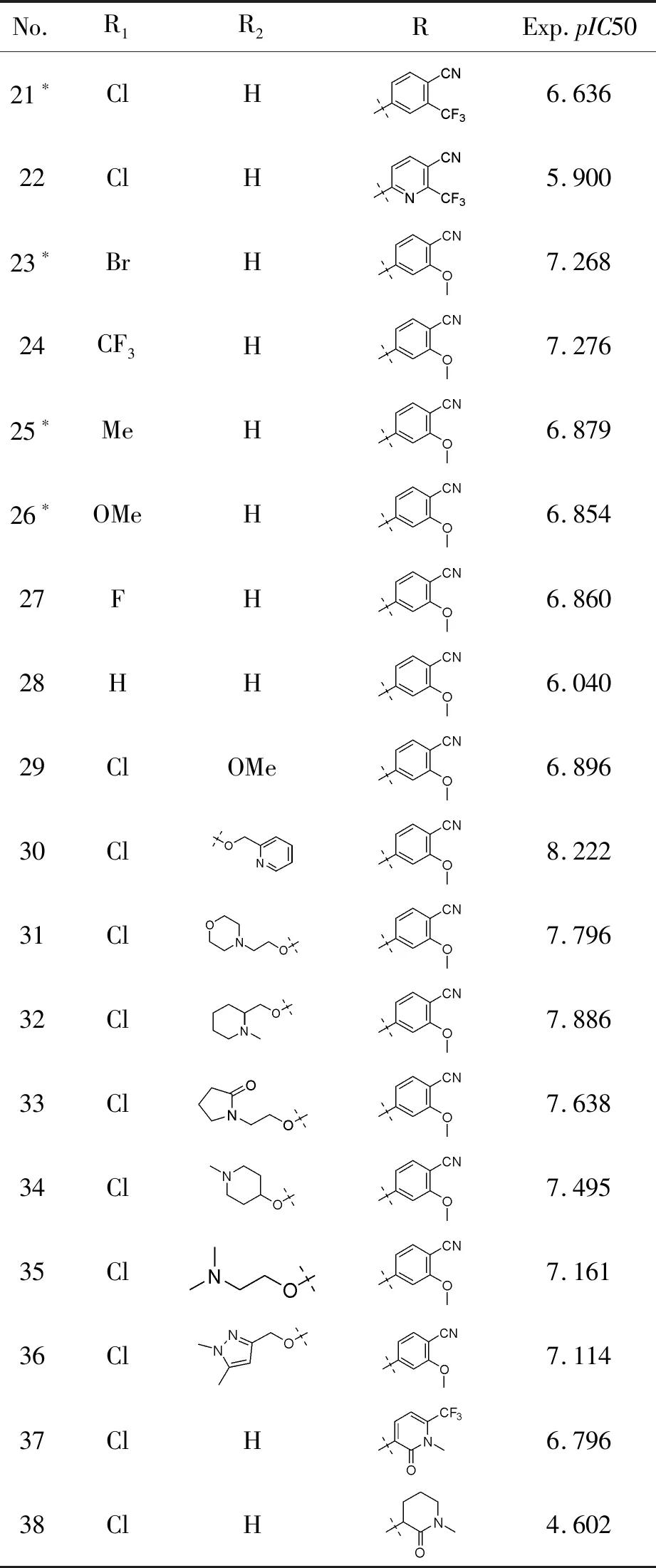
续表(表1)
1.2 3D-QSAR模型的建立
将能量最小的分子在Sybyl-X 2.0软件中进行公共骨架的分子叠合,这是构建3D-QSAR模型中最重要的一步[24]。在文章中,由于化合物No.30具有最高的活性,因而将其作为模板,对所有化合物进行叠合,化合物No.30和所有分子叠合图如图1、2所示。

图1 模板化合物No.30结构图
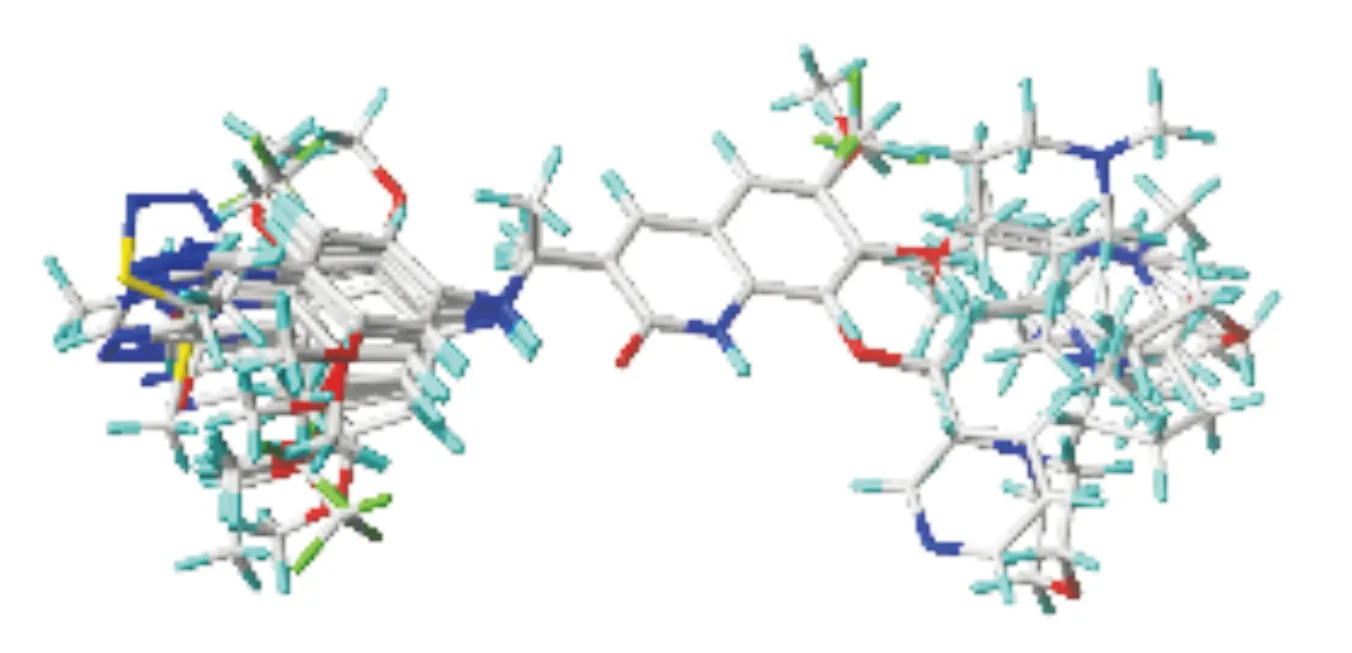
图2 模板化合物No.30与所有分子的叠合图
在SYBYL-X 2.0软件中构建了mIDH1抑制剂的CoMFA和CoMSIA模型,阐明化合物的生物活性与三维构象之间的关系[25]。CoMSIA模型不仅拥有CoMFA模型所拥有的分子场,还在其基础上增加了3个分子场,分别是氢键受体场、供体场和疏水场。不同描述符之间相互影响,为了找寻最佳交叉验证系数的分子场,需要计算不同分子场的组合来实现。进行留一法(LOO)交叉验证得到内部交叉验证系数(Q2)以及最佳主成分数N,并以非交叉验证方法计算出模型的非交叉验证相关系数(r2)、F(the fischer ratio)统计值和标准偏差(SEE)。评价3D-QSAR模型的外部预测相关系数用下式计算[26-27]:
(1)
式中:SD为测试集中的化合物与训练集中的化合物的平均抑制活性偏差的平方和;PRESS为实际抑制活性与预测活性偏差的平方和。
1.3 分子对接
首先,从蛋白数据库(protein data bank,PDB)中获取蛋白IDH1(PDB ID:6O2Y,分辨率是2.80Å)的晶体结构,在sybyl软件中对该蛋白进行一系列的非常关键的准备工作,如加氢、加电荷、删除配体等。其次,使用sybyl对小分子与蛋白进行对接,在对接过程中使用Surflex-Dock模块,Surflex-Dock(SFXC)为关键的对接模式,其他参数保持默认[28]。
2 结果和讨论
2.1 3D-QSAR结果和分析
随机选择30个化合物作为训练集构建CoMFA和CoMSIA模型,剩余的化合物作为测试集被用于检验模型的准确可靠性。所得到的相关统计参数如表2所示。一般认为,Q2>0.5,表明该模型是可靠的并且预测能力较好[29]。由表可知,CoMFA 模型的N=3,Q2=0.754(>0.5),R2=0.946,SEE=0.211,从模型中看出,2个场均对活性值产生影响,立体场的贡献是0.437,静电场的贡献是0.563,从贡献值可以看出立体场中的空间效应的影响小于静电场中的电性分布的影响。CoMSIA模型得到的结果是Q2=0.545、R2=0.955、SEE=0.196、N=4、F=132.466。立体场的贡献为0.198,静电场的贡献为0.612,疏水场的贡献为0.190。由于在CoMSIA模型中考虑了疏水场,因此所建立的CoMSAI-ESH模型是可靠的,并具有良好预测性的能力。将训练集和测试集的实验pIC50与预测pIC50的数值用散点图对其进行线性回归。所得拟合曲线如图3所示,所有化合物的预测活性都在趋势线附近,表明该化合物的实际活性与预测活性非常吻合。数据表明,3D-QSAR模型(CoMFA和CoMSIA)具有良好的稳定性和可靠的预测能力。
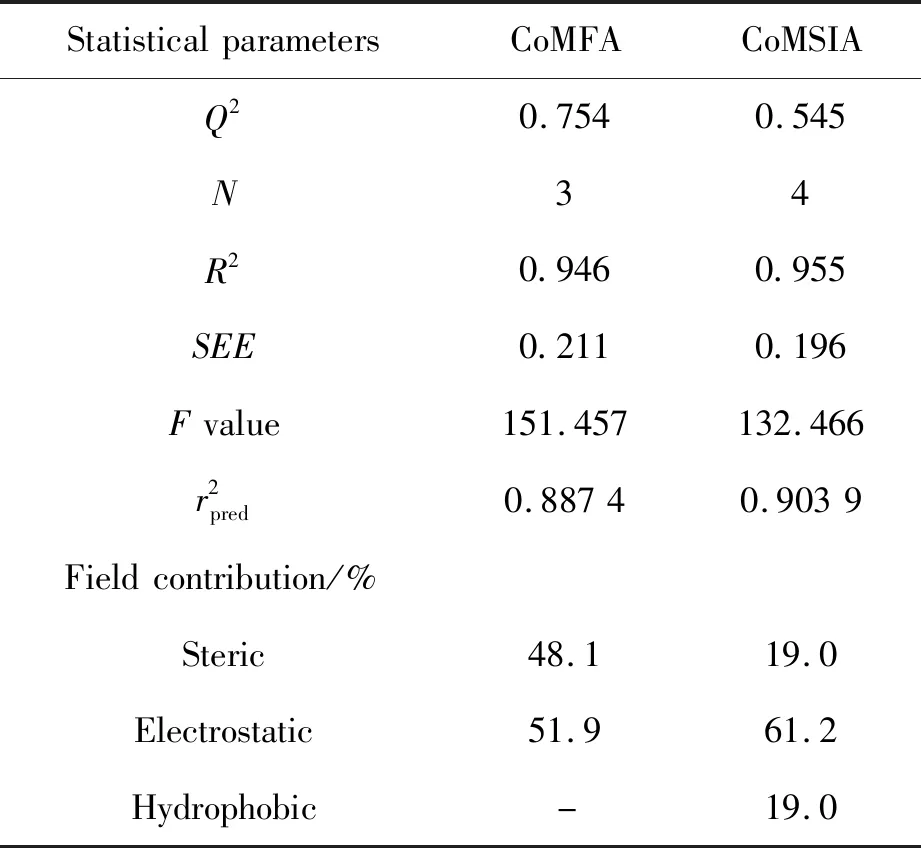
表2 CoMFA 和CoMSIA 模型的统计学数据
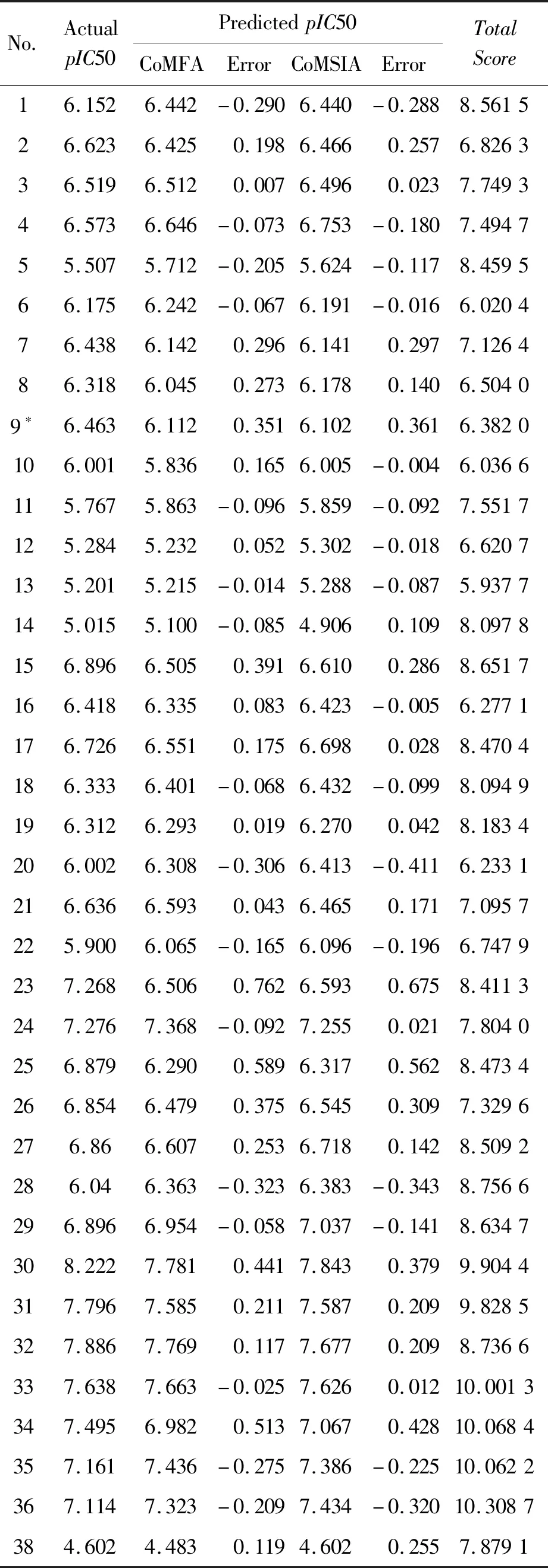
表3 CoMFA 和CoMSIA 中训练集与测试集化合物的pIC50实验值和预测值及其误差

图3 CoMFA和CoMSIA中训练集与测试集化合物的pIC50实验值和预测值之间的拟合曲线
2.2 CoMFA和CoMSIA等势图分析
使用实际活性最高的的化合物30作为模板进行CoMFA与CoMSIA的三维等势图,进而分析化合物结构与活性的关系。图4为CoMFA模型的立体场与静电场等势图。在立体场中(图4(a)),绿色区域显示提高生物活性的有效方法为引入体积较大的基团,而黄色区域则相反。从图4中可以看出,7位主要被绿色覆盖,这说明在此处引入体积大的基团,化合物的活性会明显提高。通过观察化合物15,发现在7位没有取代基取代,之后观察发现化合物20在第7位有取代基取代,发现化合物20的活性高于化合物15的活性。观察R取代基,发现此区域中主要被黄色块覆盖,表明在此区域引入体积大的基团会影响化合物活性,化合物01(pIC50=8.508)的R取代基比化合物05的R基大,故化合物01(pIC50=6.152)的活性比化合物05的大(pIC50=5.507)。在CoMFA静电等势图中,增加化合物的活性的方法为在红色区域引入负电性基团及在蓝色区域引入正电性基团。通过观察图4(b)可知,R取代基区域有蓝色存在,提高活性的方法为加入正电性的基团。发现在活性方面,化合物9优于化合物6,由于在化合物9(pIC50=6.418)的3位有羟基化合物取代而化合物6(pIC50=6.175)无取代。在立体电势与静电电势方面,CoMSIA模型与CoMFA模型比较相似。黄色区域的存在说明在该位置被疏水基团取代能增强化合物的活性,灰色区域则相反。图5(c)中可以看出,7位取代基上的部分灰色图表明,增强此化合物的活性的有效方法为在此区域引入疏水基团。R取代基的苯环3位上也有一部分绿色图块,在后面的新化合物设计中可以适当将氧甲基换成亲水基团,如羟基或氨基等。R取代基上主要是黄色区域,此处应更多考虑疏水性基团。
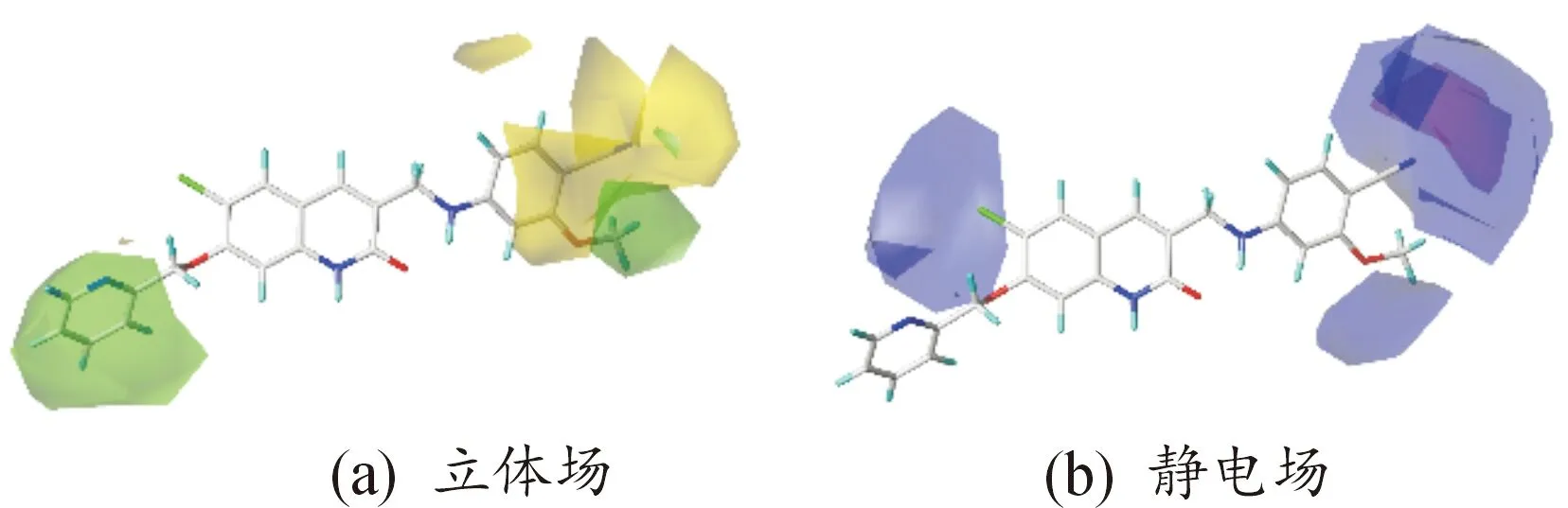
图4 CoMFA模型等势图
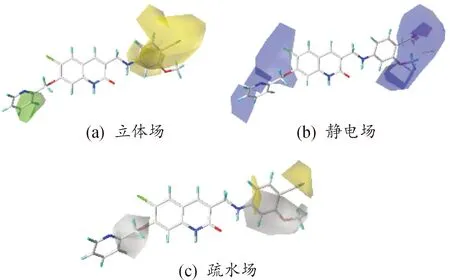
图5 CoMSIA模型等势图
2.3 分子对接结果与分析
为了检验分子对接的可靠性,将原始配体提取出来重新对接(图6)。图6中黄色表示重新对接后的构象,原始配体用绿色表示。很明显,2个配体分子的对接构象与其实验构象叠合得很好,且RMSD值为0.515 9Å(<2.0 Å)[30],说明文章采用的对接方式是可靠的。
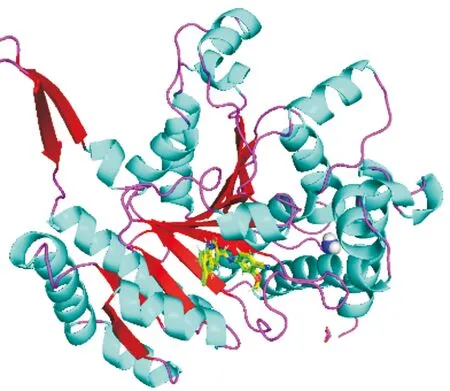
图6 原始配体的重新对接图
38个喹啉酮类化合物与靶标蛋白的结合位点对接图见图7。对接结果打分最高的是化合物No.36(TotalScore=10.308 7),No.36呈“V”形,在由Arg109、Ala111、Leu120、Trp124、Ile128、Ile130、Val255、Ala258、Met259、Trp267、Asp279、Val281、Ala282和Tyr285形成的结合口袋中。

图7 所有化合物位于IDH1蛋白的结合位点对接图
从图8(a)中可以看出,氰基与Leu120的骨架酰胺NH形成氢键而取代基苄胺(NH)与Ile128的羰基形成H键。母核喹啉酮的6位取代基氯原子与Met259的硫原子形成氢键,并填充由残基Trp267,Ala258,Trp124和Val281形成的疏水口袋。母环喹啉酮的羰基(-C=O)和氮上的H原子分别与氨基酸残基Arg109和Asp279的氧原子形成氢键。残基Trp267与母核的苯环形成π-π相互作用力。这和以前的文献报道有相似之处[15-16]。氢键与疏水作用的存在,有助于小分子化合物与靶标蛋白酶的结合,进而提高抑制剂的生物活性。而打分最低的化合物No.22(TotalScore=5.937 7)只与靶标蛋白氨基酸残基Arg109、Met259和Asp279形成氢键和与Trp267形成π-π相互作用力。说明R2取代基应考虑分子量大的基团,R基团适当增加一些亲水基团。这与QSAR的结果一致。从图8(b)中可知,No.30(TotalScore=9.044)呈“S”字结合在由以上氨基酸残基形成的结合口袋中。与No.36不同的是,氨基酸残基Cys269的磺酸基与R2取代基的苯环形成了一个相互作用力,这也使得No.30呈“S”形,抑制作用降低。这为后续设计新化合物提供了重要的信息。
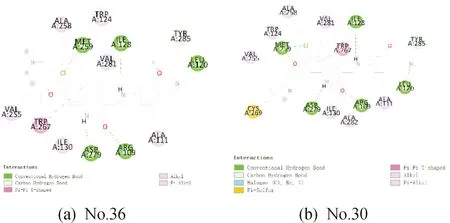
图8 化合物No.36和No.30与蛋白相互作用二维图
2.4 新化合物的设计和活性预测
通过CoMFA和 CoMSIA模型基于相同的结构和等势图的相关分析可知,在苯环的6位上应该增加正电性基团,氨基取代基上应该增加疏水性基团,找寻更多与受体的疏水相互作用。在本文中,采用化合物中活性最高的分子No.30作为模板对mIDH1抑制剂进行优化设计,得到8个新的mIDH1抑制剂分子,其预测活性与对接分数均高于模板分子。结果表明,新化合物的抑制效果优于化合物30。其中化合物No.a2的活性较高。使用前期建立的3D-QSAR模型对新设计的化合物活性进行预测,预测值如表4所示。之后使用新设计的化合物与蛋白进行分子对接,分析对接结果后发现,所有新设计的化合物的对接模式与化合物30的对接模式相同。然而对于化合物a8的基团修饰并未很好地提高化合物与靶标结合的对接分数,而化合物a1、a2、a4和a6的结构改造不仅抑制活性比No.30高,而且与蛋白质结合的对接分数也很高。然而对于化合物a8的基团修饰并未很好地提高化合物与靶标结合的对接分数,而化合物a1、a2、a4和a6的结构改造不仅得到的抑制活性比No.30高,而且与蛋白质结合的对接分数也很高。

表4 新化合物和模板分子的结构及预测活性值
2.5 ADME/T性质和合成可行性预测
为了计算新化合物的ADME/T性质,利用在线服务器pkCSM(http://biosig.unimelb.edu.au/pkcsm/)[31]评估化合物的成药性,相关参数见表5。对于新化合物合成的难易程度,运用SwissADME(http://www.swissadme.ch/index.php)[32]在线预测,如表6所示。
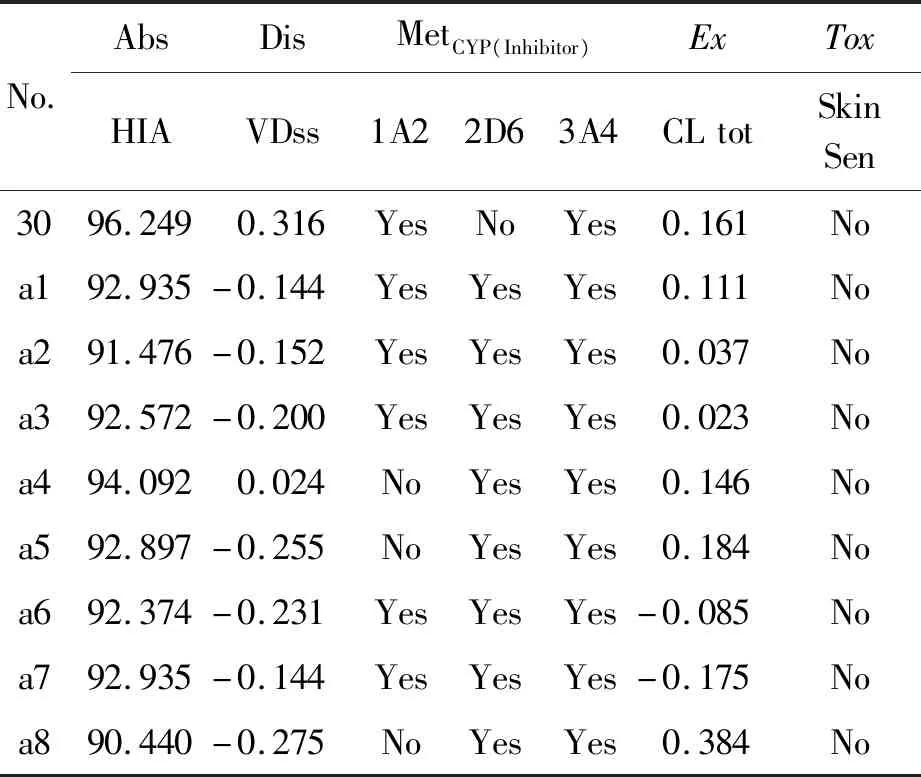
表5 新化合物及模板分子类五原则
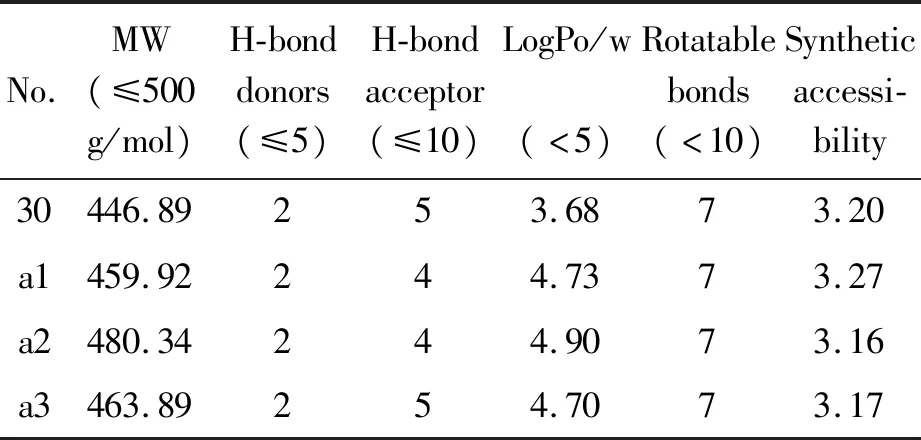
表6 No.30及其衍生物的ADME/T性质(Lipinski类五原则)

续表(表6)
从表5、6中可以看出,新设计的化合物,其肠道吸收率(Human intestinal absorption)为90.440%至94.092(>30%),表明所有新化合物均具有高吸收性能,尤其是对于化合物a1,a4和a7。新设计的化合物在体内均能被CYP450亚型2D6和3A4抑制剂代谢,而a4和a8对于CYP450 1A2亚型不会被代谢。根据预测的总清除率,所有化合物均可被肾脏和肝组织共同清除,也不会引起皮肤过敏。除了a6不符合分子类五原则以外,其他化合物均有潜力成为新型IDH1抑制剂。
3 结论
IDH1是一种备受关注的靶点。在研究中,建立了3D-QSAR模型,从而揭示了化合物结构与活性之间的关系,依据化合物与分子的对接信息,分析受体与配体之间的结合,设计并预测了8种新化合物。利用等势图更好地分析化合物结构和活性之间的关系。通过分析等势图获取了影响这一系列化合物活性的关键结构,表明新药设计可以通过在7位增加取代基的体积来增加化合物的活性,以及在此处引入疏水性基团,还可以通过在化合物的3 位引入羟基化合物的取代基,以及R取代基处有更多的疏水性基团取代。根据不同基团取代对化合物进行改造从而设计活性更高的新化合物。此外,文章还发现氢键和疏水键及静电相互作用有助于配体与受体的结合。化合物大多可以与靶标蛋白的关键氨基酸残基Arg109和Asp279等形成氢键,这可以使化合物与活性口袋更好地结合,氢键的存在对增加抑制剂的活性发挥着积极作用。本文获得了8个新颖且具有潜在IDH1抑制活性的含喹啉酮化合物,具有良好的可行性和ADMET评价结果。综上所述,通过三维定量构效关系模型的构建、验证和分析,结合分子对接机制的分析,可为后续开发新的药物提供新思路和新方向。
