催化裂化反应热计算方法的研究
2022-01-12王龙延宋业恒陈曼桥唐娉玺王宝石
王龙延,宋业恒,陈曼桥,唐娉玺,经 铁,王宝石
(中石化炼化工程(集团)股份有限公司洛阳技术研发中心,河南 洛阳 471003)
反应热直接影响着流化催化裂化装置(FCCU)反应-再生系统的热平衡,是工业FCCU工程设计和运行标定中热平衡核算和基础能耗计算的重要参数。反应热测算方法包括热力学法(或称为产品分析法,基于燃烧热或生成热)、热平衡实测法,以及分子膨胀法、催化焦法和二参数关联法等经验式法[1-2]。
受基础数据精准测定困难、关联式建立时所用原料油和催化剂与当前实际情况不同等因素所限,目前主要采用工业FCCU反应-再生系统热平衡来计算已有工业装置反应热,用分子膨胀法来测算尚未工业应用的新工艺技术的反应热。反应-再生系统热平衡法计算反应热需对FCCU进行准确的技术标定,该方法无法测算新工艺过程的反应热。分子膨胀法可用中试装置的详细物料平衡数据和产品性质数据进行计算,但该方法对缩合生焦、氢转移和异构化等二次反应没有充分考虑。
反应热不仅与FCCU原料油组成、生产方案和操作条件相关,还与反应工艺和催化剂性能有密切关系。过去的20多年,为满足生产清洁油品和多产低碳烯烃的市场需求,新催化材料和新工艺技术广泛应用,也带来工艺参数的相应变化。此外,原料范围变得越来越宽泛,加氢处理过的原料也越来越多。这些变化使采用经验式法计算反应热时的参数取值失去了背景。本课题在分析现有催化裂化反应热测算方法的基础上,提出了用典型反应计算催化裂化反应热的方法(简称典型反应法)。该方法把FCCU反应器中发生的反应划分为裂解、脱氢、氢转移、异构化、H2S生成和缩合生焦6类典型化学反应,利用每类典型反应的热效应数据和反应数量来计算催化裂化的反应热。用该方法计算了6套大型工业FCCU的反应热数据,并与分子膨胀法、催化焦法和反应-再生系统热平衡法的计算结果进行对比。
1 典型反应法计算催化裂化反应热
1.1 催化裂化的典型化学反应分类
按照重质烃催化裂化化学反应的特征,把FCCU反应器中发生的重要的化学反应(包括一次反应和二次反应)分为6类:(a)产物比原料物质的量增加、相对分子质量减小的分子裂解反应;(b)产物比原料物质的量增加、相对分子质量减小的分子脱氢反应;(c)产物物质的量和相对分子质量与原料相比基本不变的双分子氢转移反应;(d)产物物质的量和相对分子质量与原料相比基本不变的单分子异构化反应;(e)产物物质的量和相对分子质量与原料相比有增有减的缩合生焦、烯烃环化脱氢及芳环缩合等反应;(f)含硫化合物转化为H2S的反应。
1.2 典型反应的热效应数据
文献[2]依据典型化合物的生成热数据、键能数据和形成碳正离子能量数据计算出了裂解、异构化、环化、脱氢和氢转移反应的热效应数据。文献[3]给出的链烷烃环化脱氢反应的热效应。据文献[4]的结论,H2S中的硫大约有2/3来自非噻吩类硫化物,1/3来自噻吩类硫化物,可以从键能数据算出催化裂化反应中H2S生成反应的反应热。若把缩合生焦反应作为持续的环化脱氢和芳环缩合来处理,根据纽银林等[5]和宋海涛等[6]的研究结果,焦炭的氢碳质量比在0.05~0.10之间,其可萃取焦的多环芳烃的环数在3~8之间,烷基侧链碳数不大于8。由此,从键能数据可大致算出缩合生焦的典型反应热数据。相关反应热数据(平均值)列于表1。
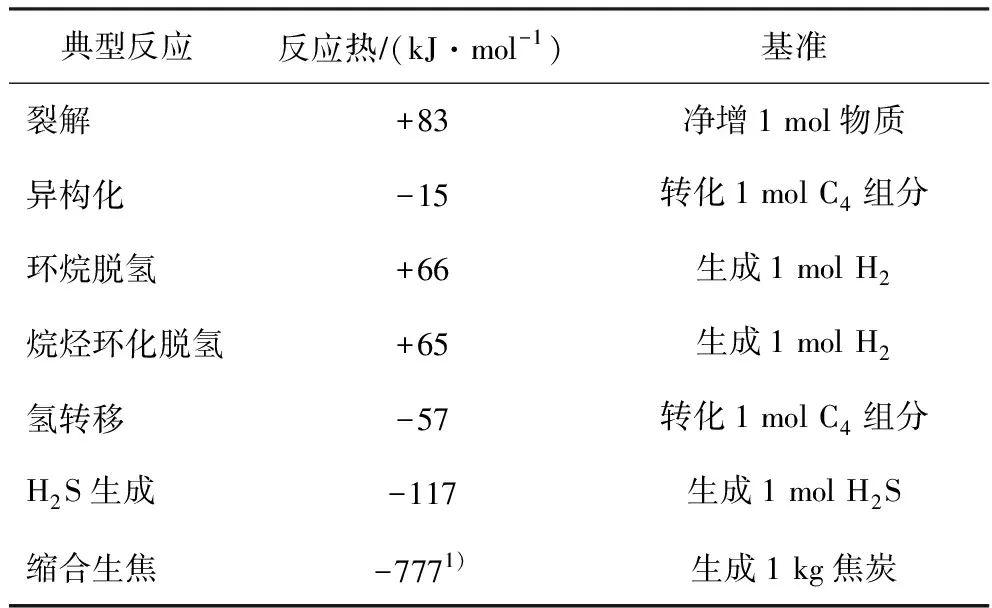
表1 不同类型反应的典型热效应数据
1.3 典型反应的反应数量计算
以1 mol 原料为计算基准。FCCU反应器中实际发生裂解反应(a)的净增物质的量na为:
na=∑(Yi×MF/Mi)-1
(1)
式中:Yi为反应产物中组分i(不包括焦炭、H2和H2S)的产率,%;MF和Mi分别为原料油和组分i的摩尔质量,g/mol。
烃分子脱氢反应(b)以及烯烃环化脱氢和芳环缩合生焦反应(e)伴随的脱氢反应所生产的H2,共同构成了FCCU反应器最终H2产物,其催化裂化过程生成H2的物质的量nb为:
nb=YH2×MF/MH2
(2)
式中:YH2为H2的产率,%;MH2为H2的摩尔质量,g/mol。
参照John[7]的研究结果,可以大致计算发生氢转移反应的C4组分的物质的量nHT(C4)。
(3)
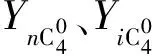
假设液体产物组分发生氢转移反应的量与C4组分发生氢转移反应的量相当,则发生氢转移反应组分的总物质的量nc为:
nc=nHT(C4)×[1+(YGL/MGL+YLCO/MLCO+
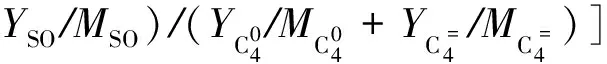
(4)
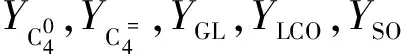
在典型的FCCU操作条件下,只有烯烃的异构化反应(双键转移、顺-反结构异构和支链异构反应)才能对反应热有实质性影响。根据John[7]和Buchanan[8]的研究结果,可以大致计算出发生异构化反应的C4组分的物质的量nIS(C4)。
(5)
式中,YiC4为异丁烯的产率,%。
假设液体产物组分发生异构化反应的量与C4组分发生异构化反应的量相当,则发生异构化反应组分的总物质的量nd为:
nd=nIS(C4)×[1+(YGL/MGL+YLCO/MLCO+
(6)
缩合生焦反应(e)最终生成了高度缩合的焦炭,可用FCCU焦炭产率来计算缩合生成焦炭的质量。
me=MF×YCK/1 000
(7)
式中:me为1 mol原料生成焦炭的质量,kg;YCK为焦炭产率,%。
对于杂原子化合物的转化反应(f),因这类反应数量占比较小且全面计算比较困难,本课题仅考虑H2S生成反应。催化裂化过程生成H2S的物质的量nf为:
nf=YH2S×MF/MH2S
(8)
式中:YH2S为H2S的产率,%;MH2S为H2S的摩尔质量,g/mol。
1.4 催化裂化反应热的计算式
用典型反应法计算催化裂化反应热的方法是根据FCCU反应器中上述6类化学反应实际发生的数量和该类型反应的典型热效应数据来计算催化裂化反应热的方法。以典型化学反应的反应热来计算催化裂化总反应热的计算式为:
ΔHR=∑(ΔHj×nj)
(9)
式中:ΔHR为催化裂化反应热,kJ/mol;ΔHj为上述6类典型反应中第j类反应的反应热,kJ/mol;nj为1 mol原料发生第j类典型反应的物质的量。根据式(9)还可以得到单位质量原料的催化裂化反应热。
QR=1 000×ΔHR/MF
(10)
式中,QR为催化裂化反应热,kJ/kg。
由上述内容可以看出,当原料组成与性质一定时,只要细物料平衡数据和产品性质相同,无论采用何种催化剂和工艺参数,采用典型反应法计算出的催化裂化反应热数据是恒定的。从这一点看,符合热力学状态函数的属性。
2 反应热计算示例与讨论
2.1 计算结果
表2是根据6套大型FCCU的标定结果[9-10],采用典型反应法计算出的反应热数据,以及与采用分子膨胀法、催化焦法和热平衡测算法计算出的反应热数据的对比。
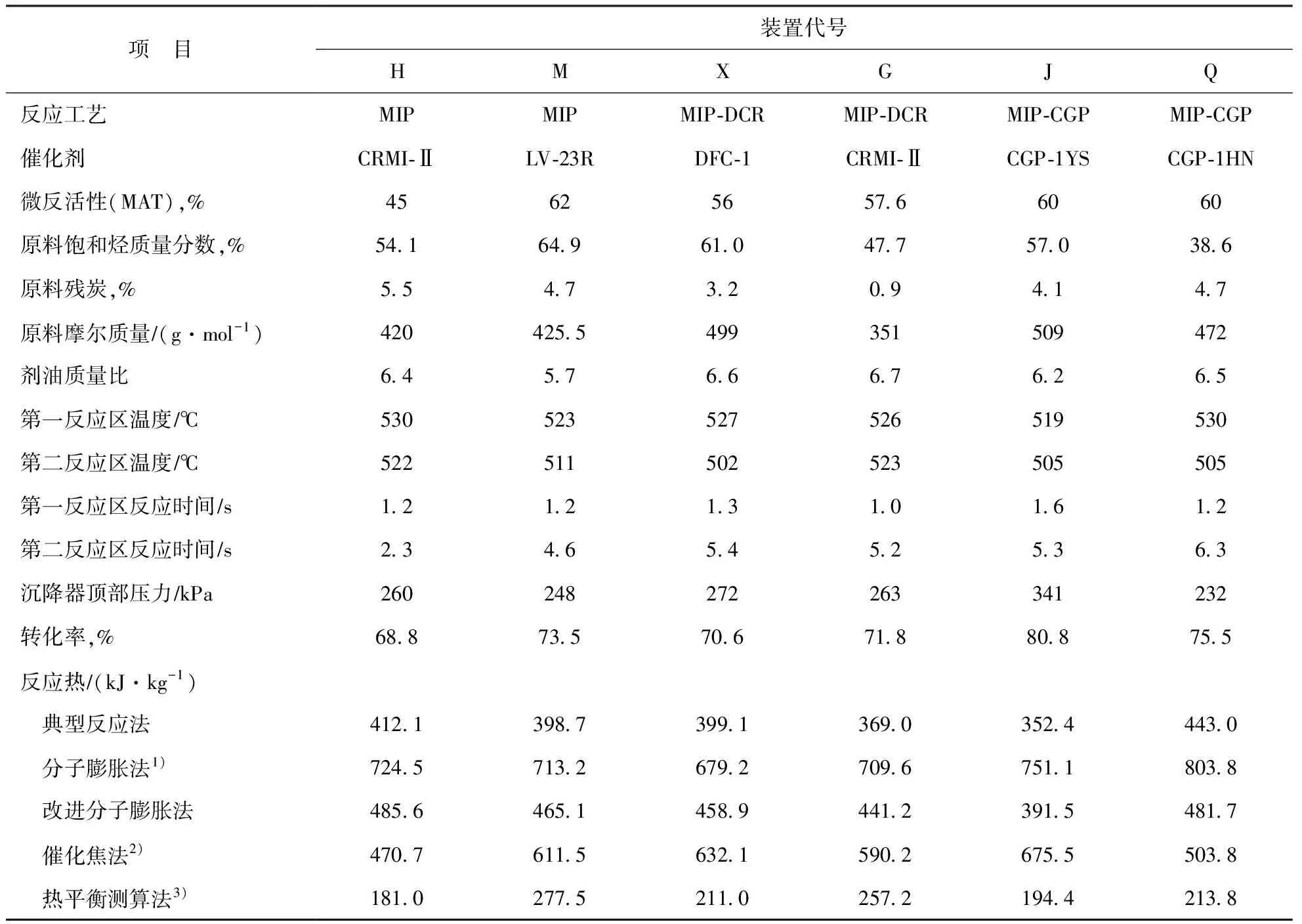
表2 催化裂化反应热计算结果
由表2可以看出,采用典型反应法计算出的6套装置反应热的算术平均值为395.7 kJ/kg。而采用分子膨胀法、催化焦法和热平衡法时对应的数据分别是730.2,580.6,222.5 kJ/kg。尽管上述6套装置的原料油、催化剂和反应条件有所差异,但同为MIP同系工艺技术,典型反应法计算出的反应热数据与装置反应-再生系统热平衡法数据最为接近。从式(1)~式(10)可以看出,采用典型反应法计算的反应热与原料油和油浆的摩尔质量数据密切相关,准确地测定原料和油浆的摩尔质量是该方法的基础。例如,G装置因掺炼7%焦化汽油,其原料的平均摩尔质量只有351 g/mol,实际上重油反应热数据可能更高一些。
2.2 典型反应法与经验式法的比较
典型反应法计算的反应热数值普遍低于经验式法,就工业装置而言,平均比分子膨胀法低45.8%,比催化焦法低31.8%。可以理解为,变径流化床反应器的MIP同系工艺使氢转移和异构化等放热反应得以强化,反应热降低了38%左右。典型反应法考虑了催化裂化二次反应,计算结果体现了反应本质变化,而经验式法却无法反映出这种变化。
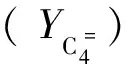
(11)
把式(11)代入分子膨胀法的反应热计算式中取得的一组反应热数据(称为改进分子膨胀法)也列入表2。此结果表明,虽然改进的分子膨胀法能够反映氢转移反应对反应热的贡献(例如J装置氢转移系数较高),但总体上看,釆用改进分子膨胀法计算的6套大型工业MIP同系工艺装置的反应热数值比原方法降低了37.8%,降低幅度比采用典型反应法时(降幅45.8%)小一些,从一个方面证明了改进分子膨胀法只考虑了氢转移反应因素,没有考虑异构化反应因素。
2.3 典型反应法与热平衡法的比较
由表2可算出,采用典型反应法比采用热平衡法得到的反应热数据平均高出了77.8%。准确多次对FCCU进行系统的测试标定是反应-再生系统热衡算取得反应热数据的基础[1]。实际上,工业生产装置要做到两次以上的原料性质不变、反应-再生系统和产品分离系统操作参数不发生波动、产品供应相对稳定的技术标定几乎不可能。工业生产装置标定期间涉及的参数计量、样品采集和分析化验环节众多,每一个环节的任何参数出现误差都会造成反应热数据的失真。
热平衡法用反应-再生系统热衡算来倒推计算催化裂化的催化剂循环量和剂油比数据,计算过程中假定焦炭吸附热等于焦炭脱附热(也可以不计算吸附热和脱附热),由此得到反应器热衡算和反应-再生系统热衡算反应热不相等的结果。这是导致热平衡法与其他方法计算结果差距较大的原因之一。以X装置为例,通过反应-再生系统热衡算得知,反应热只占焦炭燃烧放热量的6%。可见,其他项目累积的热量测算误差为3%时就会导致反应热计算值的相对误差达到50%。理论上讲,如果仅对反应器系统(从催化剂预提升到待生剂汽提)进行热衡算,计量和分析范围大幅减少,核算精度将提高。对X装置的反应系统热衡算结果表明,反应热占催化剂带入到反应系统热量的15.5%,若其他项目累积热量测算误差为3%,其反应热计算值的相对误差则降到20%以内。
反应系统热衡算不可避免要用到焦炭的吸附热数据。理论上讲,因为温度不同,反应器中的焦炭吸附热要大于再生器中焦炭脱附热。对焦炭吸附热分别取值3 373,2 350,0 kJ/kg,并采用碳差法计算X装置的催化剂循环量(2 182 t/h)和剂油质量比(7.12),对该装置反应系统按照再生催化剂降温放热和焦炭吸附放热、反应物料(含提升干气和终止剂)与蒸汽升温汽化吸热,以及反应系统散热这3大项进行热衡算,计算出来的反应热分别为806,710,504 kJ/kg。由这些数据可见,采用不同范围的热衡算方法得到的结果差别很大,用工业标定数据通过反应-再生系统热平衡法计算的反应热也未必比其他方法更接近真实值。可见,对测算反应热数据的工业装置热平衡方法还需要进一步深入研究,加以改进和完善。
典型反应法采用某些反应的典型数据来代替全部同类反应实际数据,且对于异构化、氢转移和生焦反应等放热反应热效应的研究还有待进一步量化,因此该方法计算的反应热数据不一定十分准确可靠。但该方法相对简单易行,计算所需要的基础数据主要是详细物料平衡数据和各产品的摩尔质量。因此,该方法对于中试装置和工业装置核算都能快捷实现。
典型反应法计算的反应热与经验式法相对接近,其基本原理符合近年来对催化裂化技术研究所取得的新认识规律,也基本符合反应热的热力学状态函数属性,可作为FCCU新工艺、新材料和新设备研究试验的定性评价和方向性分析,当然也可以通过大量中试装置试验数据和工业装置热平衡法测定数据关联,从而取得该方法与热平衡法相对吻合的数据。
3 结 论
(1)典型反应法利用催化裂化典型反应的热效应数据和反应数量计算催化裂化的反应热,基本原理符合反应热的热力学状态函数属性,是催化裂化反应热计算方法的尝试。
(2)与分子膨胀法、催化焦法计算结果相比,典型反应法因考虑了氢转移和异构化等二次反应放热,计算得到的反应热平均比分子膨胀法和催化焦法分别低45.8%和31.8%。
(3)典型反应法与反应-再生系统热平衡法相比,反应热计算结果高出77.8%。其原因一方面是典型反应未必能真正代表整个复杂反应体系,另一方面是反应-再生系统热平衡法也存在很大误差。
(4)如果按照碳差法计算催化剂循环量,并用催化剂在反应系统降温放热数据对反应系统热衡算得到的反应热数值要远远高于反应-再生系统热平衡法,也明显高于典型反应法。
