How non-rapid eye movement sleep and Alzheimer pathology are linked
2022-01-11AnneliesFalterMaartenVanDenBossche
Annelies Falter,Maarten J A Van Den Bossche
Annelies Falter,Maarten J A Van Den Bossche,Department of Geriatric Psychiatry,University Psychiatric Center KU Leuven,Leuven 3000,Belgium
Maarten J A Van Den Bossche,Center for Neuropsychiatry,Research Group Psychiatry,Department of Neurosciences,Leuven Brain Institute,KU Leuven,Leuven 3000,Belgium
Abstract Alzheimer's disease (AD) is a multifactorial neurodegenerative disorder characterized by the presence of senile plaques and neurofibrillary tangles.Research attempts to identify characteristic factors that are associated with the presence of the AD pathology on the one hand and that increase the risk of developing AD on the other.Changes in non-rapid eye movement (NREM) sleep may meet both requirements for various reasons.First,NREM-sleep is important for optimal memory function.In addition,studies report that the presence of AD pathology is associated with NREM-sleep changes.Finally,more and more results appear to suggest that sleep problems are not only a symptom of AD but can also increase the risk of AD.Several of these studies suggest that it is primarily a lack of NREM-sleep that is responsible for this increased risk.However,the majority investigated sleep only through subjective reporting,as a result of which NREMsleep could not be analyzed separately.The aim of this literature study is therefore to present the results of the studies that relate the AD pathology and NREM-sleep (registered by electroencephalography).Furthermore,we try to evaluate whether NREM-sleep analysis could be used to support the diagnosis of AD and whether NREM-sleep deficiency could be a causal factor in the development of AD.
Key Words:Alzheimer’s disease;Mild cognitive impairment;Sleep;Non-rapid eye movement sleep;Amyloid beta-peptides;Tau proteins;Electroencephalography
INTRODUCTION
Alzheimer’s disease
Worldwide,approximately 50 million people suffer from dementia.The most common cause of dementia is Alzheimer's disease (AD)[1].The number of people affected by this neurodegenerative disease is increasing every year.The WHO considers AD as a priority because it is one of the major causes of morbidity and mortality in the aging population.
Pathogenesis
The two main pathological abnormalities found in AD are amyloid plaques and neurofibrillary tangles.Familial forms of AD exist in which hereditary mutations lead to the development of the disease.However,sporadic occurrence is much more common.Amyloid plaques accumulate extracellularly.They are largely composed of amyloid-beta-42 (Aβ42) and to a lesser extent of amyloid-beta 40 (Aβ40),both products of amyloid precursor protein (APP) metabolism.Aβ42 is less soluble than Aβ40.Generally,they are first detected in the neocortex.The subcortical structures are affected at a later stage[2,3].The neurofibrillary tangles consist of paired phosphorylated tau-helices.These tangles accumulate in the neuronal cytoplasm and this damages the neurons.In contrast to plaques,the accumulation of neurofibrillary tangles is more predictable.Tau pathology typically begins in the medial temporal lobe allocortex (MTL;consisting of the hippocampal region,perirhinal,entorhinal,and parahippocampal cortices),where amyloid plaques accumulate less frequently,and then spread to the associative neocortex.Neuronal loss and synaptic damage usually develop after the tangles have formed[2].
Although now sometimes challenged,the most accepted hypothesis is still that the disease process is triggered by an increased production of the pathological Aβ peptides from APP by β-and γ-secretase,and by a reduced elimination of these pathological peptides (the amyloid hypothesis).According to this hypothesis,tangle formation and neuronal dysfunction follow downstream of the plaques.However,there is a strong association between the clinical symptoms,neurodegeneration and the tangles,and tau can also induce neurodegeneration independently of Aβ.It could therefore also be possible that tau and Aβ exert their effectviaother pathways and amplify each other's harmful effects[4].Both seem essential in the pathogenesis of AD,but it is still unclear how they precisely affect each other.While the current hypothesis is that the accumulation of these proteins is causally related to synaptic and neuronal loss in the brain,AD is considered a complex and multifactorial disorder,in which both genetic and environmental factors affect the development and course of the disease[4].
Clinical manifestations
Progressive problems with episodic memory are usually the first complaint of AD patients.In an early stage,mainly short-term memory is involved.However,as AD progresses,other cognitive domains are also affected.Often topographic disorientation,problems with word-finding,sentence comprehension and behavioral problems will follow[2].When there is an objective memory impairment without affecting the other cognitive domains and with a limited impact on daily functioning,this is called amnestic mild cognitive impairment (aMCI).This is seen as an intermediate stage,as individuals with aMCI have a strong increased risk of progressing to AD[5].
Although memory complaints are usually the first symptom that alerts both the environment and the patient to AD,sleep problems often precede these by several years.However,problems with sleep without the presence of memory complaints are often not immediately associated with AD.As the disease progresses,the severity of these sleep problems grows,and this is accompanied by a major impact on the quality of life of both the patient and the people around.
In addition,sleep problems are no longer only considered to be a major symptom of AD,but also a factor that may adversely affect the pathogenesis and symptomatology of this neurodegenerative disease.Episodic memory impairment is indeed the main characteristic of AD,but sleep,independent of AD,is essential for the optimal functioning of this cognitive domain.
Non-rapid eye movement-sleep
Sleep,based on electroencephalogram (EEG) oscillations,is divided into non-rapid eye movement (NREM)1-,NREM2-,NREM3-and REM-sleep.During a sleep cycle,different phases occur.One night has multiple cycles.Two characteristics of REMsleep are low amplitude desynchronized fast waves and muscle atony.NREM1 is a short transition phase between awake and sleep in which alpha waves (10-15 Hz)decrease while theta waves (4-8 Hz) become predominant.Sleep spindles (11-15 Hz)and K-complexes (KC;<1 Hz) that occur on a background of theta waves are typical of NREM2.Spindles (11-15 Hz) are generated in the thalamus and synchronized by thalamo-cortical interactions[6,7].KC are thought to protect sleep from external stimuli.During NREM3 (also called slow-wave sleep;SWS),delta waves,waves with low frequency (0.5-4 Hz) and high amplitude,are predominant along with slow oscillations (SO),slow waves (<1.25 Hz) with high amplitude[8].The activity of delta waves and SO together is sometimes called slow-wave activity (SWA).The SO arise from the superficial layers of the frontal cortex.Spindles are also present during NREM3.Sharp wave ripples (SWR) (80-140 Hz),which can be described as flares of synchronized neuronal activity in the hippocampus,are a third feature of NREM3.
For storing information in the long-term memory,NREM-sleep in particular seems to be important.To ensure a transfer of information,the neurons that represent a specific memory in the hippocampus are repeatedly activated.If the neocortex is activated simultaneously,the transfer of information takes place[9].This co-activation gives the slow-learning cortex the opportunity to integrate the new information into the long-term memory.This happens mainly during NREM3.Especially SWR,spindles and SO appear to be essential for this process.A recent study[10] concluded that it is the exact timing and precise coupling of SWR,spindles and SO that ensure the transfer of information from the hippocampus to the cortex.The SWR precedes the spindle and activates the neuronal sequence associated with a recent memory.The spindles and the subsequent cortical SO help process this memory.The timing of these events determines the quality of the information transfer that takes place during the SWS[8].
NREM-sleep,normal aging and AD
Aging is associated with physiological changes in sleep patterns.In general,older people sleep less,go to bed earlier,get up earlier,sleep more during the day,wake up more at night and have difficulties in falling asleep again[11].Age-related changes in the oscillations can also be observed on the EEG.Both the quantity and quality of NREM3 decreases while the length of NREM1 in particular and to a lesser extent NREM2 increases[12].Older people have a decreased percentage of SWA and the amplitude is on average lower compared to younger people.This difference is most pronounced in the frontal brain regions where the SWA originates[13].The density,duration and peak and mean amplitude of sleep spindles also decrease with aging[14,15].
As early initiation of therapy usually yields better results,attempts have been made to identify symptoms that are specific to AD and that can be measured early in the course of the disease.The previous paragraph indicates that sleep quantity and quality decreases with aging even in the absence of neurodegenerative pathology,and are therefore not unique to AD.Nevertheless,interest in sleep in AD has increased rapidly in recent years.This is because,on the one hand,specific sleep problems such as excessive daytime sleepiness and insomnia are more prevalent in AD.On the other hand,these sleep problems,similar to the formation of senile plaques and neurofibrillary tangles,seem to appear several years before the cognitive decline.As AD is considered a multifactorial disease,another focus of current research is identifying risk factors.Sleep deprivation (SD;no sleep for one night) and Alzheimer's pathology such as senile plaques and neurofibrillary tangles have been shown to be associated.Thus,there may be a bi-directional relationship between sleep problems and AD.
As NREM-sleep is of great importance for proper functioning of episodic memory,which is also the cognitive domain that often deteriorates first in AD,several studies have concluded that NREM-sleep in particular is responsible for the association found between AD-pathology and sleep.However,the majority of the studies on this topic analyzed sleep through subjective reporting and did not differentiate between the different sleep stages.The purpose of this literature review is therefore to provide an overview of the literature investigating the relationship between NREM-sleep and AD in humans.In addition,studies will be discussed that specifically investigated the relationship between AD-pathology (in particular amyloid and tau) and NREM-sleep and which analyzed the possible causative role of NREM-sleep in the pathogenesis of AD.Subsequently,we briefly review the experiments that investigated whether NREM-sleep might be one of the pathways through which AD-pathology affects memory.Finally,based on the results found,it is discussed whether measuring NREM-sleepviaEEG could in the future be used in the prevention,diagnosis and treatment of AD.
SEARCH METHODS
We performed a search of the literature using the PubMed,Embase and Cochrane Library databases.The search terms used at the start of the literature study were“Alzheimer's disease” AND “NREM” OR “non-rapid eye movement” OR “nonREM”OR “SWS” OR “slow-wave”.
English-language studies in which human sleep was objectively measured by EEG and which included aMCI or AD as the study population were included.Studies analyzing the sleep of subjects without cognitive impairment were included only if the purpose of these studies was to gain more insight into the association between AD and NREM-sleep,and if amyloid-beta or tau measurements were compared with NREMEEG data.The references of the above articles were also searched for additional articles that met the inclusion criteria.
RESULTS
One hundred and twenty-nine individual studies were found and screened,of which 31 met our inclusion criteria.An overview of the article selection is shown schematically in Figure 1.The results found are further described below.
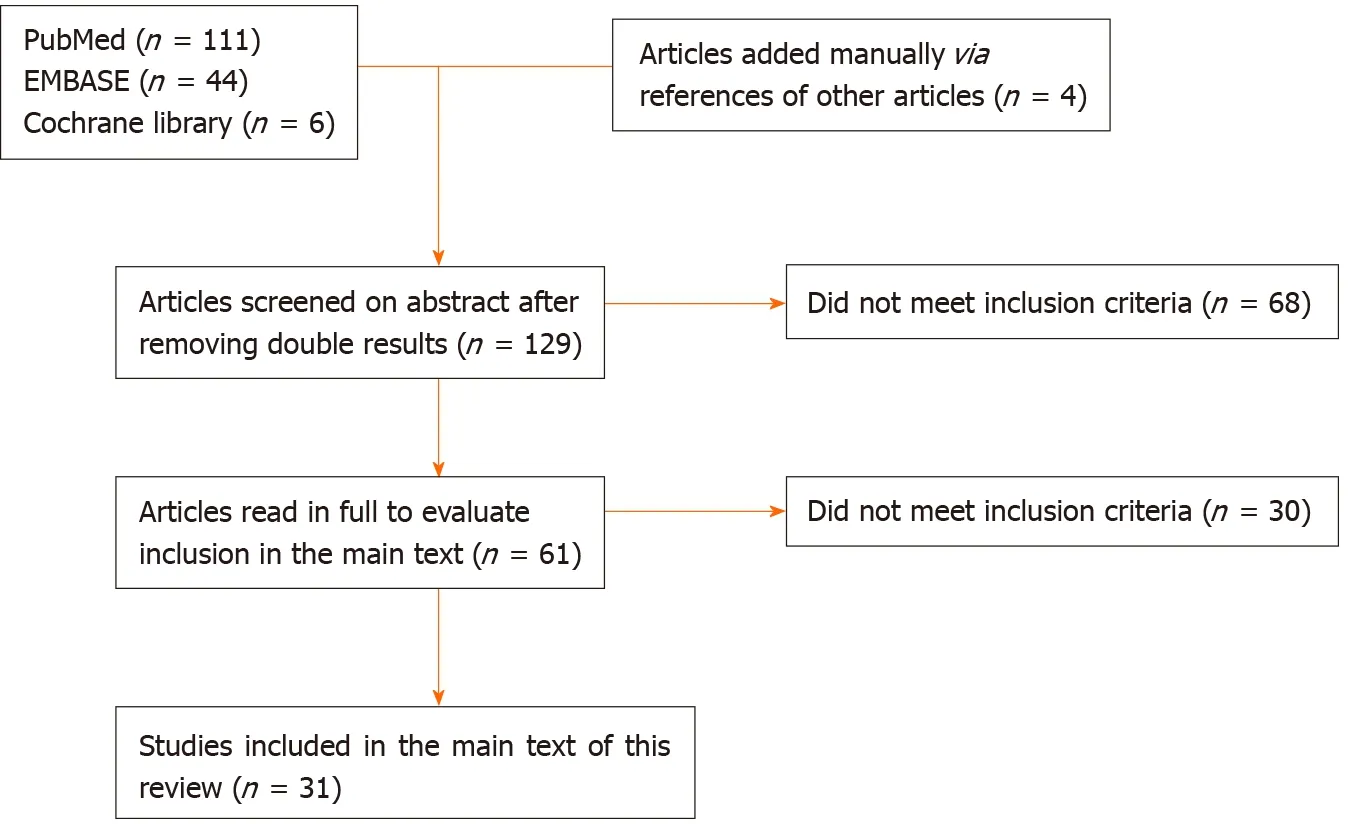
Figure 1 Schematic overview of article selection.
NREM-sleep changes in AD and aMCI cognitive impairment
Prinzet al[16] and Vitielloet al[17] were among the first to detect that the percentage of NREM (duration NREM/duration total sleep) was significantly lower in AD patients than in elderly with intact cognition (EIC).More recent studies[6,18,19] also confirmed that people suffering from AD have a lower percentage of NREM3 compared to EIC.In several studies[6,18,20,21],a smaller percentage of NREM-sleep was associated with lower MMSE scores,not only in the total study group but also within the AD group.As diagnostic markers that can contribute to early identification of AD are sought after,Westerberget al[22] studied NREM in aMCI patients.They found that the percentage of NREM3 was less in the aMCI group than in EIC.Within this aMCI group,patients with a lower percentage of NREM-sleep scored worse on a test in which they were asked to reproduce in the morning what they had studied the night before[22].Although the mean percentage of NREM3-sleep in aMCI patients in another study[19] was lower than in the EIC group,this difference was not significant.This study by Gorgoniet al[19] also included AD patients and they found a significant difference in NREM3-sleep percentage between AD patients and EIC.The findings by Redaet al[20] were similar:The difference in percentage of NREM3 was not significant between aMCI and AD,but significant between AD and the EIC group.In contrast to Gorgoniet al[19],the difference between aMCI and the EIC group in this study did however reach the significance level[20].A smaller study[23],where we have to be careful when interpreting the results due to the heterogeneity of the different types of dementia that were included,analyzed the SWA within the 0.4-3.6 Hz range,the activity that predominates during NREM3 sleep.A shift was found within this frequency spectrum in patients with dementia.There was a significant decrease in the percentage and incidence of delta waves with a frequency less than 1.6 Hz in the group with dementia.At the same time,there was a significant increase in the incidence of delta waves with a frequency higher than 2 Hz[23].
Many functions of NREM-sleep and its microstructures are not yet known.What is known is that SWR,SO,spindles and KC,that mainly occur during NREM-sleep,are important for our memory function.Therefore,these microstructures of NREM-sleep were also investigated in patients with aMCI or AD.Rauchset al[14] hypothesized that patients with AD would have fewer spindles compared to EIC,but this difference was not significant.In a more in-depth analysis whereby a distinction was made between fast (13-15 Hz) and slow (11-13 Hz) spindles[9],only the amount of fast spindles was significantly reduced in AD patients[14].Westerberget al[22] made a distinction based on location,and studied the spindles separately in a parietal and a frontal lead,where the fast and slow spindles,respectively,are most pronounced[7].They noted a significant reduction in the aMCI group of the fast spindles in the frontal but not in the parietal lead.For the slow spindles,there was no difference in both leads[22].The reduction of fast spindles was confirmed by Gorgoniet al[19].They found a significant reduction in density (spindle number/length NREM sleep) of all spindles and of the fast spindles registeredviathe parietal lead in aMCI and AD,while the slow spindles did not differ.There was no difference between aMCI and AD[19].A recent study[18]analyzed the total amount of spindles during NREM-sleep.The density,amplitude and duration of these spindles were lower in AD than in aMCI and EIC.They were also lower in aMCI compared to EIC.Fast spindle density and spindle amplitude were both positively correlated with the MMSE scores[18].Within the aMCI and AD subgroups,the occurrence of the spindles was positively correlated with the performance on a memory test in which the studied material was examined in the morning[19,22].
De Gennaroet al[6] also analyzed the KC.AD patients had a drastic decrease (40%)in KC-density during NREM-sleep.This decrease was most pronounced in the frontal lead[6].Redaet al[20] reported similar observations.Patients with AD had a lower KCdensity.The density was significantly less not only compared with EIC,but also with the aMCI group.The difference was not significant between aMCI patients and EIC[20].A recent study[18] measured the frontal KC-density in EIC,aMCI and AD.There was no significant difference in density between EIC and aMCI,while the density and amplitude were significantly lower in AD.Only the KC-amplitude differed between aMCI and EIC.No difference was found within the three groups in the duration of the KC[18].The first prospective cohort study[24] on KC-complexes in aMCI confirms most of these results.In this study,the sleep of both aMCI and EIC was recorded after specific time intervals during a follow-up period of 2 years.Subjects who developed AD and subjects whose aMCI status remained stable during two years of follow-up,were in the analysis divided in a progressive aMCI group (pMCI) and a stable aMCI group (sMCI),respectively.The KC-density at baseline and at 6 mo did not significantly differ among the three groups.In contrast,the KC-amplitude was significantly lower in sMCI and pMCI compared with EIC,while there was no difference between sMCI and pMCI subjects.At 12 and 24 mo,the KC-density and KCamplitude was higher in EIC compared with sMCI and pMCI,but was also higher in sMCI than in pMCI.At 12 mo,when the pMCI subjects did not yet meet the criteria of AD,the KC-density and KC-amplitude was nevertheless already significantly lower in pMCI than in sMCI.The KC-density correlated positively with the MMSE scores in all studies[6,18,24].
The link between NREM-sleep and Aβ and tau
Aβ42 and tau are pivotal in the pathogenesis of AD.Therefore,it was also investigated whether specific NREM EEG-abnormalities could reflect the presymptomatic presence of Aβ and tau in the cerebrospinal fluid (CSF) or on imaging.
Aβ-and tau-specific positron emission tomography scans and the NREM-sleep EEG
Manderet al[25] detected a relationship between Aβ deposition in the medial prefrontal cortex and a reduction in NREM3 in EIC.A detailed analysis showed that this association was due to an underlying association between Aβ and less activity in the 0.6-1 Hz range.This inverse relationship was strongest between Aβ in the medial prefrontal cortex and sleep recordedviathe frontal EEG lead.There was no significant association with the total 1-4 Hz frequency spectrum[25].
Luceyet al[26] analyzed not only Aβ but also tau presence on the brain scans of older adults who were predominantly cognitively intact (some subjects with aMCI).The percentage of 1-4 Hz-SWA decreased when more Aβ and tau were seen on imaging.For Aβ,there was an inverse association between Aβ deposition in the frontal,temporal and parietal cortices and both the 1-2 Hz and 1-4 Hz activity.The total amount of tau,and tau at the level of the entorhinal,parahippocampal and inferior temporal areas,was in these regions most strongly associated with the 1-2 Hz activity.The associations found with tau were much stronger than those with Aβ,and the associations with Aβ disappeared after statistical correction for multiple testing[26].In both studies[25,26],the associations found remained significant when corrected for cortical thickness.Wineret al[10] tried to determine whether there are EEG abnormalities that are only associated with Aβ and not with tau and vice versa.The MTL and the cortex were analyzed because these regions are particularly susceptible to tau-and Aβ-damage in AD,respectively[2].They observed that a weaker coupling between SO and spindles was associated with more accumulation of tau in the MTL[10].There was no association between the strength of the coupling and cortical Aβ.However,for Aβ,they found,in line with the results of Manderet al[25],a significant inverse correlation between cortical Aβ and activity in the 0-6-1 HZ range[10].A reduction in 0.6-1 Hz-SWA predicted a higher amount of cortical Aβ.MTL-tau had no significant association with this frequency interval or with any other subgroup frequency of the 0.6-4 Hz range[10].A new study of Wineret al[27] aimed to address the lack of longitudinal data on this topic.The sleep of EIC was registered during one night in the beginning of the study.Around the same time positron emission tomography (PET) scans were taken in order to analyze Aβ accumulation.During a follow-up period of on average 3.7 years,multiple follow-up PET scans were performed to measure the change in Aβ.They found a negative correlation between the proportion of 0.6-1 Hz SWA at baseline and the change in Aβ during the follow-up.This negative correlation was frequency range specific,as they did not find an association between total SWA (0.8-4.6 Hz) and Aβ change.In addition,the strength of SO-spindle coupling was not associated with the rate of Aβ change[27].
Aβ-and tau-CSF concentrations and the NREM-sleep-EEG
Luceyet al[26] observed no correlation between CSF-Aβ42 and total NREM-SWA while using a similar study design where imaging was replaced by CSF analysis.In this study[26],there was however a significant inverse relationship between the 1-4.5 Hz-SWA and the ratio between CSF-tau and CSF-Aβ42 (CSF-tau/CSF-Aβ42).The ratio between phosphorylated tau and CSF-Aβ42 (CSF-p-tau/CSF-Aβ42) was also analyzed separately and this relationship was also significant.The inverse relationship between CSF-tau/CSF-Aβ42 and the 1-2 Hz SWA was the strongest.Kamet al[28] explored associations with the microstructural features of NREM-sleep.A decrease in spindle density during NREM2 was correlated with an increase in CSF-tau and CSF-tau/Aβ42 in EIC.This relationship was strongest between specifically CSF-tau and fast spindle density,while there was no relationship with the slow spindles.Frontally recorded 0.5-4 Hz-SWA was also inversely related with CSF-Aβ42 but not with CSF-tau[28].In another study[29],CSF-Aβ42 was inversely related with the duration and percentage of NREM3,while there was no significant relationship between CSF-Aβ42,total sleep length and the length of the other sleep stages.Additionally,no association was found between the percentage of NREM3 and CSF-tau[29].In mild and more advanced AD patients,there was only a correlation between CSF-tau and a decreased percentage of NREM3;this association did not apply to CSF-Aβ42 and the percentage of NREM3[21].
The impact of a NREM-sleep deficit on CSF-Aβ and CSF-tau
Several studies[30,31] have reported that CSF-Aβ fluctuates over the course of a 24-h period,with the highest concentration being measured in the evening and the lowest in the morning after sleep.Following this finding,and with the aim of gaining more insight into the relationship between NREM sleep and AD pathology,the effect of sleep deprivation (SD) on the CSF-Aβ and CSF-tau concentrations of adults with intact cognition were examined.Lumbar catheters were used in two studies[32,33] to frequently monitor CSF levels during both normal sleep and SD.In the study of Oomset al[32] CSF-Aβ42 decreased significantly (6%) after sleep,while the concentration did not decrease after SD.Neither sleep nor SD significantly affected CSF-tau.Luceyet al[33] found a similar but greater increase (30%) in CSF-Aβ after SD and,in contrast to Oomset al[32],also noted an increase in CSF-tau (50%).Shokriet al[34] conducted their study assuming that radio tracers also identify soluble amyloid and therefore can show a direct increase in Aβ42 on imaging.They compared two scans of the same EIC that were taken after one night of normal sleep and after one night without sleep.SD resulted in a significant increase in Aβ42 in the right hippocampus,parahippocampus and thalamus on the second scan[34].In an attempt to determine whether NREM3 specifically contributes to this decrease in CSF-Aβ following sleep,Juet al[35]disrupted sleep only during NREM3.This intervention logically resulted in a reduced percentage of SWA and a reduced total sleep duration.The percentage of NREM1 increased,while the other sleep variables remained unchanged.After NREM3 sleep disturbance,CSF-Aβ40 increased.There was no significant association between CSF-A β40 and CSF-Aβ42 and NREM,REM,or total sleep length.No correlation was found between NREM3 disturbance and CSF-tau concentrations[35].Olssonet al[36] wanted to address the lack of knowledge on the chronic effects of SD on AD markers by allowing subjects with intact cognition to sleep only partially (max.4 h) in their sleep laboratory for five consecutive nights.In the same subjects,physiological sleep was also monitored during five other nights.There was an interval of one month between both circumstances.After the completion of each period,a lumbar puncture was performed.Their hypothesis was that five nights of partial sleep deprivation (PSD)would lead to increased CSF-Aβ or CSF-tau.However,they found no significant difference in CSF-Aβ40,CSF-Aβ42 or CSF-tau concentrations between the two conditions.EEG analysis showed that despite the shorter total sleep time (207 min) in PSD,there was no significant difference in NREM3 (4 min) length.The lengths of NREM1,NREM2 and REM were shorter with PSD (19 min,131 min and 52 min,respectively)[36].
The link between Aβ and tau,memory and NREM-sleep
Episodic memory impairment is characteristic of AD.As NREM-sleep is important for this cognitive domain,research groups have attempted to investigate whether NREMsleep is still important for memory function,when AD pathology is already present in the brain.
In the previously mentioned studies[14,19,22],there was in the total study population (EIC,aMCI and AD) a proportional relationship between the results on memory tests or MMSE scores and,among other things,the percentage of NREM3-sleep,the spindle density and the KC-density.Furthermore,this association persisted when aMCI and AD were considered separately.Liuet al[18] detected in their study the strongest associations between NREM3 percentage and MMSE,and between KCdensity and MMSE.These results are based on experiments in which the physiological sleep of aMCI,AD and EIC was measuredviaEEG during one night.A recent study[37] did more than just record physiological sleep.It used an intervention to investigate the link between NREM sleep,memory and AD.aMCI patients in this study had to study information before they went to sleep.The following night their SWA was stimulatedviaacoustic stimulation.The same experiment was repeated but without stimulation during sleep.The first observation was that acoustic stimulation significantly increased SWA.The second one was that on average aMCI patients remembered more after a night of stimulation,although the difference did not reach significance[37].The research group that previously detected the inverse correlation between cortical Aβ and SWA,also used memory tests in their experiments[25].Their hypothesis was that cortical Aβ indirectly,by reducing NREM-SWA,leads to less information stored in the cortex.Decreased SWA was in this study[25] associated with less hippocampal-cortical information transfer during NREM-sleep.Statistical analysis(structural equation models) showed that the association between cortical Aβ and impaired memory was dependent on the degree to which the 0.6-1 HZ-SWA decreased as an intermediate factor.Cortical Aβ and memory were only significantly inversely related in the statistical model in which NREM-SWA was included[25].
DISCUSSION
This review shows that there is an association between AD and NREM-sleep.The number of EEG studies examining this association is rather limited up to now,but current results suggest that NREM-sleep analysis could in the future be useful in the diagnosis,prevention and therapy of AD.
Some sleep problems,such as excessive daytime sleepiness and insomnia,are characteristic of AD.The studies discussed suggest that certain specific changes in NREM-sleep could help in the diagnosis of aMCI and AD.Based on the current literature,a decrease in the NREM3 percentage should be able to differentiate persons with AD or aMCI from EIC.Although in each study this percentage was lower in AD compared to aMCI,the difference was not always statistically significant.Nevertheless,this suggests that the percentage might decrease progressively during the course of the disease.Some microstructural properties of NREM-sleep were also found to be significantly different in AD.A reduction in fast spindle density,amplitude and duration could help to identify aMCI and AD patients.While the spindle density could not distinguish the aMCI group from the AD group in the study by Gorgoniet al[19],Liuet al[18] showed a difference in both density and amplitude between both groups.Measuring spindles and NREM3 percentage with EEG could therefore assist in diagnosis at an early stage.In contrast,KC-density was consistently lower in AD compared to the aMCI group.This parameter might therefore be especially useful at a later stage,as in the majority of experiments the KC-density was not different between aMCI and EIC.A reduction in KC-amplitude may occur earlier and might be useful in distinguishing aMCI from EIC[24].
An association between sleep and AD pathology has been demonstrated before.Aβ and tau were associated with less qualitative sleep in humans[38].However,the majority of these studies evaluated sleep subjectively and therefore could not differentiate between the different sleep stages.The discussed results suggest that AD pathology may cause EEG abnormalities particularly in NREM-sleep.Thein vivomeasurement of Aβ and tauviabrain imaging is used in the clinic to support the diagnosis of AD.Pathological abnormalities on imaging characteristic of AD cooccurred with specific EEG abnormalities during NREM-sleep in the reviewed studies.A reduced percentage of 0.6-1 Hz-SWA reflected increased cortical Aβ in both Wineret al[10] and Manderet al[25],while there was no association with tau.Due to technical limitations,Luceyet al[26] did not analyze below 1 Hz,so they could neither confirm nor refute these results.In contrast,less than 1-2 Hz SWA could indicate tau pathology in the MTL and the orbitofrontal,inferior parietal and inferior temporal cortex[26].Wineret al[10] did not confirm this association between tau and SWA,but identified a microstructural feature specifically associated with tau.The SO-spindle-coupling,and in particular the progressive loss of coupling intensity,might identify individuals with high MTL-tau earlier.The on average older population in the study by Luceyet al[26]could possibly explain the fact that they did detect an association between tau and SWA.As mentioned in the introduction,tau indeed first accumulates in the MTL and only later spreads to the cortical areas where the slow waves are generated.
An increase in the ratio of CSF-tau/CSF-Aβ42,CSF-tau,and a decrease in CSF-Aβ are also used in the clinic as biomarkers to support the diagnosis of AD[2].A preclinical rise in CSF-tau reflects an increased risk of AD.Although there is more debate about the predictive value of CSF-Aβ,the literature suggests that CSF-Aβ increases in a preclinical stage which promotes plaque formation and decreases at a later stage only when plaques are more numerous[39].These CSF biomarkers were also associated with NREM abnormalities in EIC.Reduced fast spindle density during NREM2 and 1-2 Hz-SWA were associated with an increase in CSF-p-tau,CSF-tau and CSF-tau/CSF-Aβ42[26,28].CSF-tau and CSF-p-tau are markers of neuronal damage and neurofibrillary tangles,respectively[40].These NREM changes could potentially be an early indicator of tau pathology.The results for amyloid were less consistent.Kamet al[28] and Vargaet al[29] reported an inverse association between the percentage of NREM3 and CSF-Aβ42.Luceyet al[26] could not confirm this association.Again,their study population could have some influence on the results,as there were also aMCI patients in this study,and the presence of plaques might reduce CSF-Aβ fluctuations.Hence,EEG measurements,like CSF-analysis and imaging,could be used to support the diagnosis of AD and to identify individuals at increased risk in a non-invasive way.More research is needed however,to both confirm the results discussed and to quantify these abnormalities into clinically useful parameters.
Measuring NREM-sleep might have even more clinical potential.It has been acknowledged for some time that chronic sleep problems entail an increased risk of developing AD.For example,the risk that adults with intact cognition but with untreated obstructive sleep apnea hypopnea syndrome or insomnia will develop AD later on is higher than in persons without a sleep disorder[41-43].Over the last decade,evidence is mounting that a lack of NREM3-sleep may be an important cause of this increased risk,and that NREM3 may play a causal role in the pathogenesis of AD.However,the design of the above-mentioned studies usually did not allow determination of the causality in the association found between CSF concentrations or imaging and NREM-sleep abnormalities.Additional arguments that sleep problems could potentially lead to Alzheimer's pathology are found in studies in which participants are exposed to acute SD.All SD studies[32,33,35] discussed reported a CSF-Aβ concentration that was higher after SD than after normal sleep.For tau,the effects were less consistent.In the majority of studies,no nocturnal change in CSF-tau was observed in either physiological sleep or SD.Only one study found a significant increase in CSF tau after SD[33].One possible way in which a lack of NREM3 sleep could lead to a rise in certain concentrations in the CSF is through the so-called“glymphatic system”[44].This perivascular network,found in animal models,promotes the elimination of soluble proteins,including Aβ,from the central nervous system.The glymphatic system is mainly active during sleep and its function is said to be maximal during SWS[45].The findings of Juet al[35] that a selective disruption of SWA was inversely correlated with an increase in CSF amyloid,and that there was no association with total sleep or REM sleep,are in line with this.The selective interruption of NREM3-sleep had no effect on CSF tau.This could possibly be due to the longer half-life of tau,which means that one night without sleep has a lesser effect on the clearance of tau[46].On the other hand,it is also possible that sleep has a different effect on the clearance of Aβ and tau.The fact that NREM3-sleep was ensured with PSD and that PSD did not induce an increase in CSF-Aβ42 or CSF-tau also supports the hypothesis that it is NREM3 that mainly affects CSF concentrations[36].Although it seems likely that there is an increased clearance of CSF-Aβ42 during NREM3,an increased synthesis,as suggested by Luceyet al[26],could also be partly responsible for an increase in CSF-Aβ42 after SD.A combination of reduced clearance and increased synthesis of CSF-AB42 during SD is also possible.
Studies on humans,combining EEG recording,CSF measurements and imaging over a long period of time are needed to gain more insight into the effect of NREMsleep on Aβ and tau and to determine whether the CSF fluctuations are due to increased clearance or to increased synthesis of these proteins.Despite the lack of clear understanding about the mechanism,ensuring NREM-sleep could become important in the prevention of AD.In addition,stimulation of SWS could be used to normalize CSF-Aβ and in this way reduce the risk of plaques.SWS stimulation also has potential as a treatment for the memory symptoms that characterize AD[37].Problems with the transfer of information to long-term memory are namely one of the first complaints of AD.NREM-sleep and the interaction of SO,spindles and SWR play an important role in this information transfer[8].Acoustic stimulation during SWA has previously been shown to improve memory tests in individuals with intact cognition[47].SWS stimulation was also able to improve memory in aMCI patients[37].
The hypothesis of Manderet al[25] that a reduction in SWA is one of the mechanisms by which cortical Aβ impedes the storage of information in the long-term memory,could provide a possible explanation for the observation that,despite the presence of AD pathology in an aMCI stage,SWA stimulation still had a positive effect on memory.This may explain why the decline of hippocampus-dependent memory is usually one of the first symptoms of AD,while Aβ usually does not accumulate at the hippocampus until later in the disease process.There is moreover a similarity between the regions that generate NREM-SWA and the cortical areas where Aβ usually first accumulates[48].A similar EEG study has not yet been performed in aMCI patients.However,two recent studies[49,50] in a heterogeneous group of elderly (EIC,aMCI and AD) also suggested that sleep at an early stage may partly influence the impact of cortical Aβ on memory,as the inverse relation between cortical Aβ on imaging and memory was statistically dependent on sleep continuity as an intermediate factor.Interestingly,EIC with high cortical Aβ had a more continuous sleep in this study than the EIC with little Aβ,which might be indicative of a possible protective role of sleep on cognition in the presence of high Aβ accumulation[49,50].These studies all focused on Aβ.Whether NREM-sleep disturbance is also one of the pathways through which tau affects memory is not yet known.
Qualitative NREM3-sleep seems to be essential for memory in both EIC and AD patients.Enhancement of SWA,such as by acoustic stimulation,might therefore be a potential therapy for memory problems in AD patients.
CONCLUSION
This review discussed the studies that examined the link between NREM-sleep and AD.Based on the presented results,examining NREM-sleep could be a new diagnostic tool for detecting aMCI and AD in a non-invasive way.Enhancement of NREM-sleep could be an exciting new option in the prevention and treatment of AD.Prospective and longitudinal studies that combine EEG measurements with imaging and CSF analyses are needed to gain more insight into the causality and the underlying mechanisms of this relationship.
杂志排行

World Journal of Psychiatry的其它文章
- Effectiveness of cognitive behavioral therapy-based interventions on health outcomes in patients with coronary heart disease:A metaanalysis
- How does the ‘environment’ come to the person? The ‘ecology of the person’ and addiction
- G-protein coupled receptors and synaptic plasticity in sleep deprivation
- Agmatine as a novel candidate for rapid-onset antidepressant response
- Healthy diet,depression and quality of life:A narrative review of biological mechanisms and primary prevention opportunities
- Neurodevelopmental disorders:An innovative perspective via the response to intervention model