利用微滴数字PCR技术分析转基因大豆‘GE-J12’中外源基因的拷贝数
2022-01-10李瑞环曹英芳兰青阔
赵 新 刘 双 刘 娜 李瑞环 曹英芳 兰青阔 王 永*
(1.天津市农业科学院 生物技术研究所,天津 300384;2.河北农业大学 食品科技学院,河北 保定 071001)
伴随转基因技术的发展,转基因作物及其衍生食品的安全性问题受到社会强烈的关注,国家监管部门对转基因作物的上市和推广均制定了明确的政策和法规,并对其开展严格的生物安全评价。载体全序列分析、目的基因整合和外源插入序列表达等分子特征是转基因生物安全评价的重要组成内容之一,其中转基因作物中插入外源基因的拷贝数鉴定是关键参数。外源基因整合进入受体基因组的位置和拷贝数会影响目的基因的表达和遗传稳定性,而插入基因的拷贝数相对其插入位点而言影响更为重要。当外源基因以低拷贝数(1~2个)整合到受体基因组时,通常能够稳定高效转录表达,而多拷贝数的整合则会造成基因不稳定表达,甚至沉默。因此,鉴定转基因作物的外源基因插入拷贝数非常重要。
目前,Southern 印迹杂交(Southern blot)和实时荧光定量PCR(Quantitative real-time PCR, qRT-PCR)是鉴定转基因作物外源基因拷贝数常用的2种分析技术, 已广泛应用。但这2种技术都存在一定缺陷,Southern blot方法操作步骤繁琐、工作量大、检测周期长、对试验人员操作技术要求较高;qRT-PCR方法需要依赖标准物质,构建标准曲线,而利用标准曲线进行定量本身就是一种并不十分精确的相对定量方法,且构建标准曲线的过程中涉及体系的摸索和优化,也耗费大量的时间和精力,因此qRT-PCR不是理想的转基因作物外源基因拷贝数的鉴定方法。微滴数字PCR(droplet-based digital PCR,ddPCR)是最新兴起的一项核酸检测技术,该技术不依赖任何校准物,无需构建标准曲线,以形成大量油包水微滴的形式对核酸进行成千上万倍稀释,然后以每个小微滴为独立的反应单元进行PCR反应,最后利用泊松分布原理,实现对核酸检测的绝对定量。在检测目的基因拷贝数时,利用目标基因与内标基因的双重反应,通过计算其比值,即可快速获得目标基因拷贝数。相比其他方法,该方法操作简便、快速高效、准确性高、重复性好,已成为检测外源基因拷贝数的首选方法。诸多研究表明,dd PCR已用于玉米和水稻等作物转基因插入拷贝数的检测。
抗除草剂转基因大豆‘GE-J12’是由中国农业科学院作物科学研究所研发的转G2-EPSPS
基因和GAT
基因抗除草剂大豆新品种。采用农杆菌介导转化法,将转G2-EPSPS
和GAT
的2个基因的表达载体DNA导入大豆受体中,通过多代筛选获得高耐草甘膦转基因大豆新品种。培育和推广耐草甘膦除草剂大豆新品种,对防除大豆田间杂草、降低生产成本,提高农民种豆积极性和种豆效益,增强我国大豆竞争力具有重要意义。目前,对于该转基因大豆新品种的外源基因插入拷贝数的数字PCR分析方法的研究尚未见报道。本研究以‘GE-J12’的G2-EPSPS
和GAT
外源插入目的基因序列、3′端转化体特异性序列为靶标,以Lectin
为内标基因,通过引物探针筛选、特异性测试分析,旨在建立微滴数字PCR拷贝数分析方法,以期为该转化体的安全评价提供参考。1 材料与方法
1.1 材料及仪器
抗除草剂转基因大豆品种‘GE-J12’,非转基因大豆品种‘Jack’,由中国农业科学院作物科学研究所研发;转基因玉米品种混合样(‘MON863’、‘MON810’、‘MON88017’、‘MON87427’、‘MON87460’、‘MON89034’、‘双抗12-5’、‘Bt176’、‘Bt11’、‘MIR604’、‘MIR162’、‘GA21’、‘DAS40278-9’、‘NK603’、‘TC1507’、‘T25’、‘3272’、‘4114’、‘5307’、‘59122’、‘C0030.3.5’、‘C0010.3.7’和‘IE09S034’每种转化体质量分数为1%,以非转基因玉米为填充物),其他转基因大豆品种混合样(‘A2704-12’、‘A5547-127’、‘CV127’、‘DAS68416-4’、‘FG72’、‘GTS40-3-2’、‘MON87705’、‘MON87769’、‘MON88302’、‘MON89788’、‘SHZD32-1’、‘73496’、‘356043’和‘305423’每种转化体质量分数为1%,以非转基因大豆为填充物),转基因油菜品种混合样(‘MON88302’、‘RF1’、‘RF2’、‘RF3’、‘Ms1’、‘Ms8’、‘Topas19/2’、‘T45’、‘oxy235’和‘73496’每种转化体质量分数为1%,以非转基因油菜为填充物),转基因水稻品种混合样(‘科丰2号’、‘科丰6号’、‘科丰8号’、‘克螟稻’、‘TT51-1’、‘T1C-19’、‘G6H1’、‘M12’每种转化体质量分数为1%,以非转基因水稻为填充物),转基因棉花品种混合样(‘COT102’、‘GHB614’、‘LLCOTTON25’、‘MON1445’、‘MON15985’、‘MON88913’、‘MON531’每种转化体质量分数为1%,以非转基因棉花为填充物),由本实验室收集保存。
1.2 试验方法
1
.2
.1
引物及探针的设计转基因大豆‘GE-J12’的外源基因插入位置在大豆3号染色体上,插入位点包含1个G2-EPSPS
和1个GAT
基因的表达框,不同外源基因片段之间以大豆基因组DNA片段相连接,见图1。本研究以抗除草剂转基因大豆‘GE-J12’的G2-EPSPS
和GAT
外源插入基因序列,T-DNA与大豆基因组连接区域的3′端转化体特异性序列为靶标,以Lectin
为内标,应用Primer Express 3.0设计引物和探针。引物和探针由上海生工生物工程有限公司合成,见表1。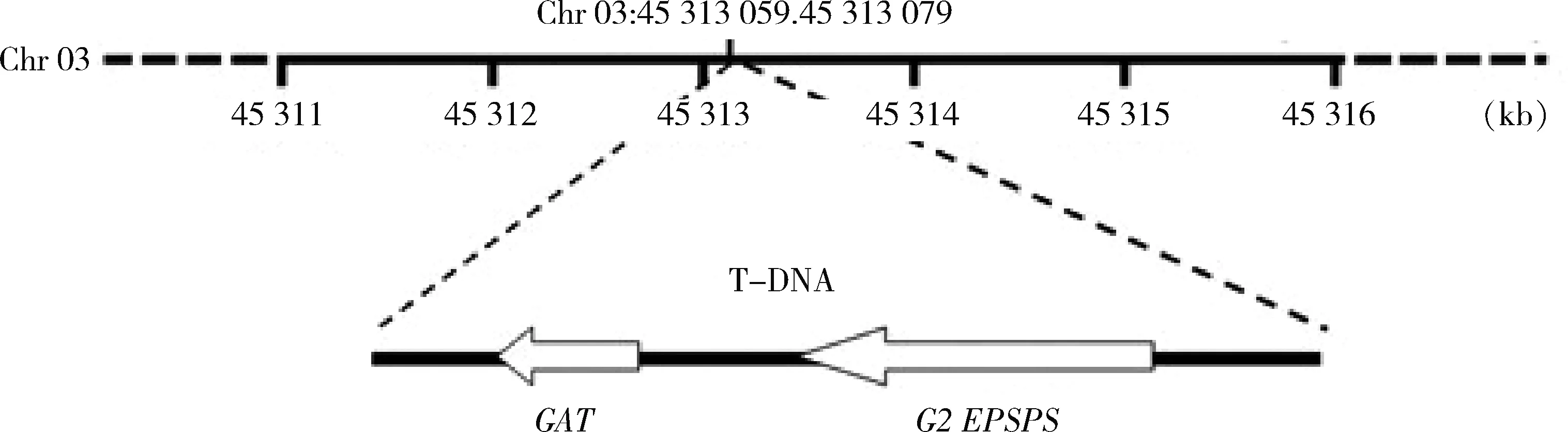
图1 抗除草剂转基因大豆‘GE-J12’外源基因插入位点
表1 PCR引物和探针序列
Table 1 PCR primers and probe sequences
名称Name引物序列(5′→3′)Primersequence(5′→3′)片段大小/bpSize序列来源SequencesourceLectin基因QF:5′-GCCCTCTACTCCACCCCCA-3′QR:5′-GCCCATCTGCAAGCCTTTTT-3′QP:5′-FAM-AGCTTCGCCGCTTCCTTCAACTTCAC-BHQ1-3′118农业部2031号公告-8-2013G2-EPSPS基因QF:5′-TCGAGATTGATGGTGGTTTGTC-3′QR:5′-TCAGCGCCACTTCAATCG-3′QP:5′-FAM-GGGCAAACTGATTTCCATAGCTT-BHQ1-3′ 90自行设计GAT基因QF:5′-GGGCAAACTGATTTCCATAGCTT-3′QR:5′-CCTCGGAGCTGGTACTGTTTCT-3′QP:5′-FAM-ATTCCACCAGGCCGAGCACTCAGA-BHQ1-3′ 81自行设计3′转化体特异性序列QF:5′-GCAGCTTGAGCTTGGATCAGA-3′QR:5′-TCATAGCGTGGTGTTTGACATAAA-3′QP:5′-FAM-TGTCGTTTCCCGCCTTCAGTTTAAACC-BHQ1-3′ 91自行设计
1
.2
.2
DNA模板的提取用TIANGEN高效植物基因组DNA提取试剂盒(天根生化科技北京有限公司),提取转基因大豆品种‘GE-J12’的DNA;非转基因大豆受体品种‘Jack’、转基因玉米混合样、其他转基因大豆混合样、转基因油菜混合样、转基因水稻混合、转基因棉花混合样,各取100 mg粉末样品,用TIANGEN高效植物基因组DNA提取试剂盒,提取DNA。用美国Bio-Rad公司的NanoDrop-100核酸蛋白定量仪测定DNA的浓度和质量,将DNA浓度统一稀释至25 ng/μL,备用。
1
.2
.3
微滴数字PCR检测体系的建立在20 μL的PCR反应体系中,包括2×ddPCR Supermixfor probes预混液10 μL,上下游引物和探针各1 μL,模板1 μL,无菌超纯水补足20 μL,混匀后离心。将其转移至微卡发生器,切忌产生气泡,同时与微卡发生器相应位置加入重油70 μL,盖上垫片,放入微滴生成器中生成微滴。吸取40 μL生成的微滴,转移至数字PCR 96孔反应板中,170 ℃热封后,在美国Bio-Rad公司的QX-100微滴数字PCR仪上开始扩增循环,PCR仪爬坡速度设置为2 ℃/s,循环参数为:95 ℃,10 min;94 ℃,30 s,60 ℃,1 min,40 个循环;98 ℃,10 min。
1
.2
.4
方法特异性检测以转基因玉米品种混合样,其他转基因大豆品种混合样,转基因油菜品种混合样,转基因水稻品种混合样,转基因棉花品种混合样和非转基因大豆品种样品的基因组DNA为模板,以去离子水作为阴性对照,按1.2.3反应程序及条件进行微滴数字PCR扩增,对检测方法的特异性进行测试,每个反应设置2个重复。
1
.2
.5
微滴数字PCR拷贝数分析以抗除草剂转基因大豆‘GE-J12’的3个单株基因组DNA为模板,以大豆内标基因Lectin
为参照,应用构建的微滴数字PCR检测体系,对3′转化体特异性序列、G2-EPSPS
和GAT
外源插入基因进行微滴数字PCR分析,测定每个基因的拷贝数浓度,每个反应设置2次重复。1
.2
.6
微滴数字PCR拷贝数计算微滴数字PCR仪自动计算每个反应的拷贝数浓度、总微滴数和阳性微滴数,总微滴数≥10 000视为有效结果,按照公式计算出试样中转基因大豆的外源目的基因在基因组插入的拷贝数。公式如下:
式中:A
,转基因大豆的外源目的基因在基因组插入的拷贝数;B
,转基因大豆的外源目的基因的拷贝数浓度,个/μL;C
,内标基因Lectin
基因的拷贝数浓度,个/μL。1
.2
.7
Southern blot方法探针制备PCR扩增使用G2-EPSPS
和GAT
基因引物,见表2,由上海生工生物工程有限公司合成。总体积为50 μL,包括10×buffer(plus Mg)预混液5 μL、dUTP标记混合物5 μL、上下游引物各1 μL、Taq酶 1 μL、1 μL质粒DNA、ddHO补足50 μL,进行PCR扩增,扩增结束后,对扩增产物进行电泳检测(1% 琼脂糖)及地高辛探针制备。根据转基因大豆‘GE-J12’转化载体全序列信息和酶切位点信息,选择常用的内切酶进行基因组 DNA酶切,酶切体系总体积为600 μL,包括 10×Buffer 60 μL,BSA 60 μL,Hin
d III/Spe I/Xba
I/Dra
I内切酶30 μL,基因组 DNA 10 μg。探针和酶切产物经电泳、转膜和杂交等后续步骤获得 Southern杂交结果。表2 Southern 杂交检测引物
Table 2 Southern blot PCR primers sequences
探针名称Name引物序列(5′→3′)Primersequence(5′→3′)片段大小/bpSize序列来源SequencesourceTP3TP3-F:GCTTCTATCGGCTGGTTTP3-R:GCCAATACGCAAACCGC902GATTP5TP5-F:ACAAACCACCATCAATCTP5-R:CGTTGTTAGCCTTGCGG724G2-EPSPS
2 结果与分析
2.1 引物和探针的特异性试验
由图2可知,只有以抗除草剂转基因大豆‘GE-J12’的基因组DNA为模板时才能获得阳性微滴和对应的拷贝数,且阳性微滴与阴性微滴界限明显,而以其他转基因作物品种混样和其他非转基因大豆品种植株受体的基因组DNA为模板均无阳性微滴,说明本方法筛选的引物探针特异性良好,可以用于检测抗除草剂转基因大豆‘GE-J12’的外源插入基因的拷贝数。
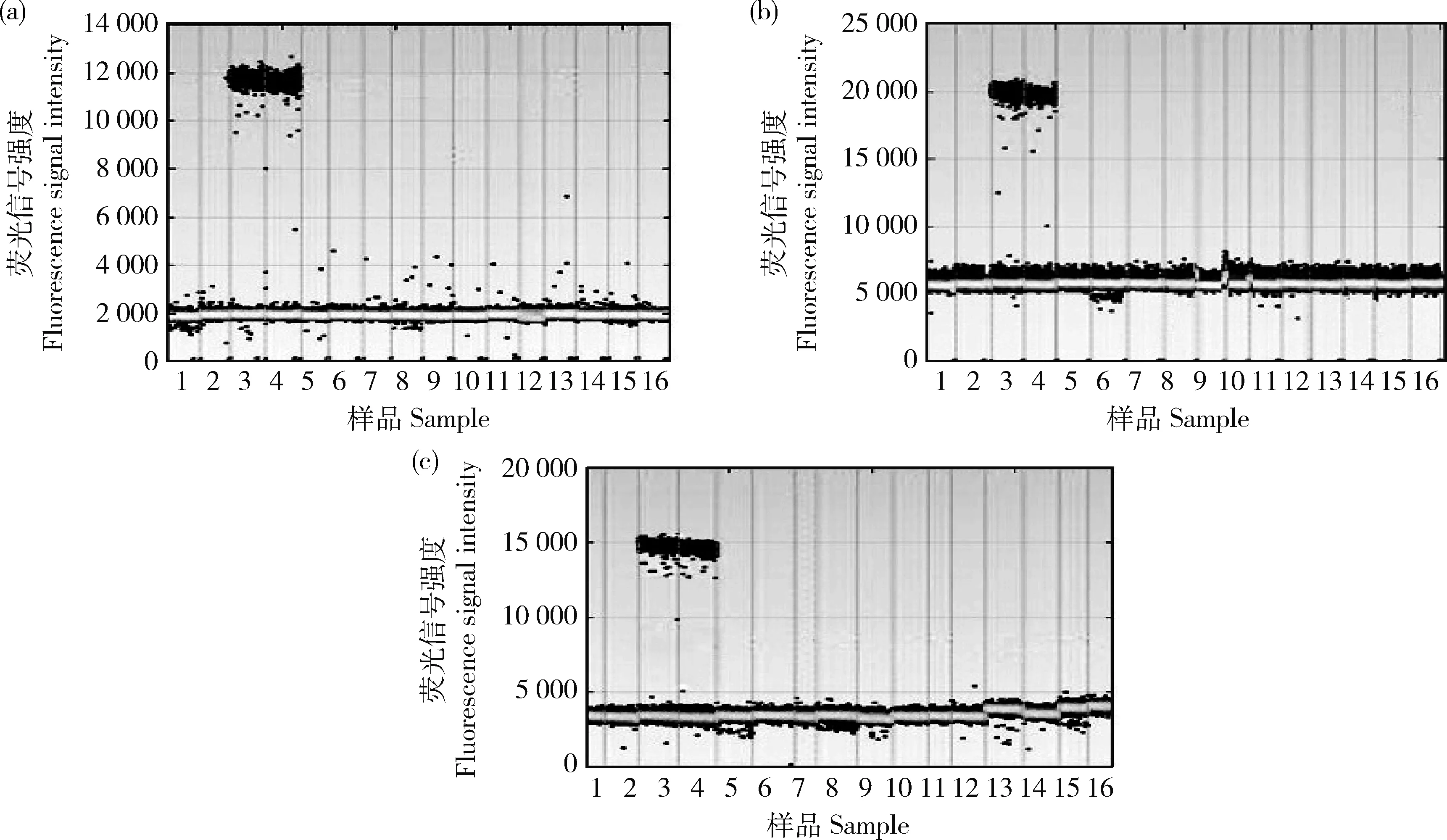
1和2,空白对照;3和4,转基因大豆‘GE-J12’;5和6,非转基因大豆(受体);7和8,转基因玉米混合样;9和10,其他转基因大豆混合样;11和12,转基因油菜混合样;13和14,转基因水稻混合样;15和16,转基因棉花混合样。
2.2 微滴数字PCR拷贝数分析
3′转化体特异性序列可在分析外源基因拷贝数的同时,通过与大豆内标基因Lectin
的比值,作为植株纯合性鉴定的引物探针,纯合阳性植株接近于1个拷贝,杂合植株接近于0.5个拷贝。由表3可知,经微滴数字PCR验证,转基因大豆品种‘GE-J12’的 3个单株中,以Lectin
作为内标,3’转化体特异性序列的拷贝数比值分别为0.96、1.03和1.02,均接近于1个拷贝,验证结果显示,转基因大豆品种‘GE-J12’的 3个单株均为纯合阳性转基因植株;3个单株外源基因G2-EPSPS
和GAT
的拷贝数比值分别为1.02、1.16、0.81和1.04、1.12、0.89,均接近于1个拷贝,参照‘GE-J12’研发者提供和全序列分析结果,验证转基因大豆‘GE-J12’的载体T-DNA在大豆基因组上为单拷贝插入,具体拷贝数浓度,见表3,微滴扩增结果,见图3、图4和图5。表3 转基因大豆‘GE-J12’数字PCR检测分析
Table 3 Analysis of digital PCR detection of transgenic soybean‘GE-J12’
基因Gene外源序列拷贝数/(个/μL)Foreignsequencecopynumber内源Lectin拷贝数/(个/μL)EndogenousLectincopynumber拷贝数比值Copynumber单株拷贝数均值Averagecopynumberperplant619.50606.501.02G2-EPSPS614.00531.501.160.99658.50813.000.81633.50606.501.04GAT595.00531.501.121.01721.00813.000.89582.00606.500.963’旁侧序列546.50531.501.031.00831.00813.001.02
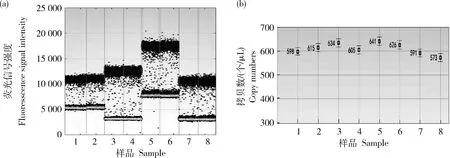
1和2,Lectin基因;3和4:G2-EPSPS基因;5和6,GAT基因;7和8,3′转化体特异序列。下同。
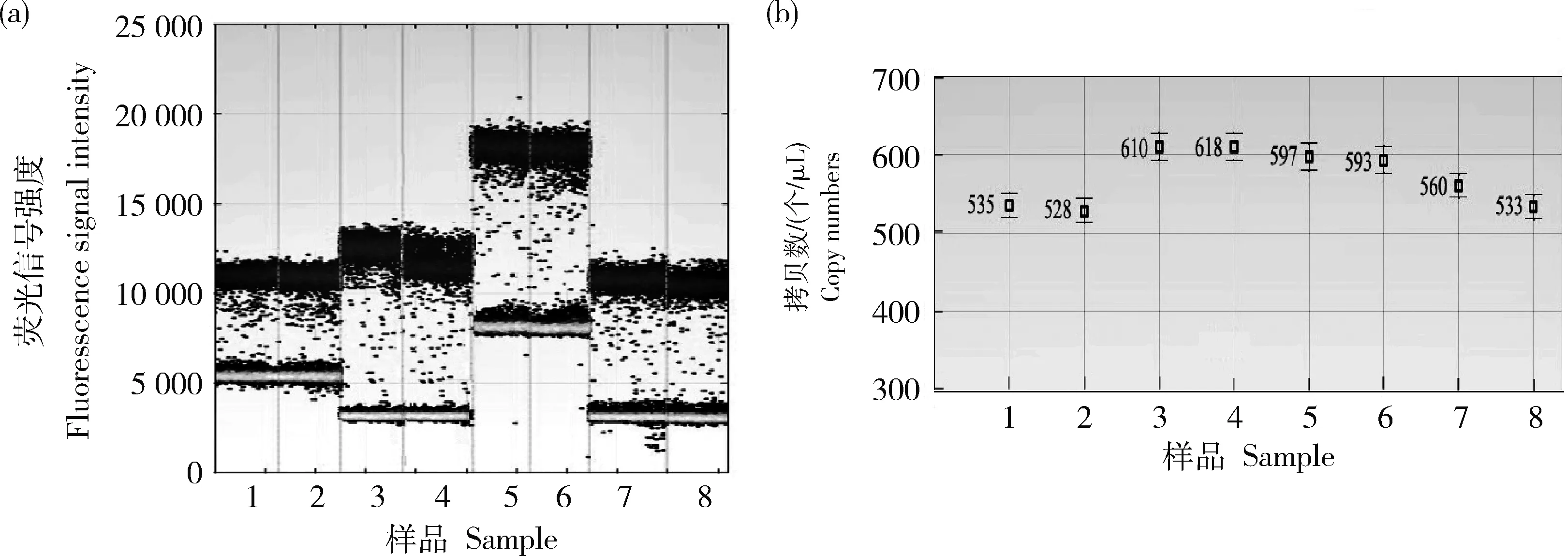
图4 转基因大豆‘GE-J12’2号单株的数字PCR扩增微滴图(a)和拷贝数浓度图(b)

图5 转基因大豆‘GE-J12’3号单株的数字PCR扩增微滴图(a)和拷贝数浓度图(b)
2.3 Southern blot结果
由图6可知,应用上述Hin
d III、Spe
I、Xba
I、Dra
I等4种内切酶酶切后转基因大豆品种‘GE-J12’和非转基因大豆品种‘Jack’的基因组DNA泳道均呈弥散状,无明显条带,说明基因组 DNA 酶切效果较好。由图7可知,用G2-EPSPS
和GAT
扩增产物为杂交探针,对酶切产物进行Southern blot检测,质粒阳性对照杂交结果正常,非转基因大豆品种(受体)‘Jack’经Hin
d III、Spe
I、Xba
I和Dra
I的酶切杂交后均未出现杂交信号,转基因大豆品种‘GE-J12’经上述4种酶切杂交后,杂交片段大小与预期大小一致(Hind III、Spe I、Xba I、Dra
I酶切后预期杂交片段大小分别为4 859、8 638、11 236和4 754 bp),且探针杂交信号均为1个,说明目的基因的拷贝数为1个。
M,1 kb DNA Ladder;1:非转基因大豆受体(Hind III),2,转基因大豆‘GE-J12’(Hind III);3,非转基因大豆受体(Spe I);4,‘GE-J12’(Spe I);5,非转基因大豆受体(Xba I);6,‘GE-J12’(Xba I);7,非转基因大豆受体(Dra I);8,‘GE-J12’(Dra I)。
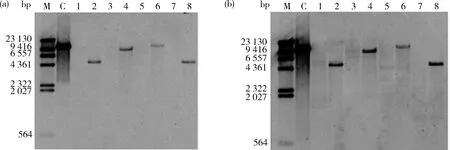
M,DNA molecular-weight marker;C,阳性质粒;1,非转基因大豆受体(Hind III);2,转基因大豆‘GE-J12’(Hind III);3,非转基因大豆受体(Spe I);4,‘GE-J12’(Spe I);5,非转基因大豆受体(XbaI);6,‘GE-J12’(XbaI);7,非转基因大豆受体(Dra I);8,‘GE-J12’(Dra I)。
3 讨 论
目前的转基因作物研究中,能够准确、快速地鉴定转基因作物中外源基因的拷贝数,已经成为转基因作物检测的关键内容。传统的Southern blot方法操作复杂、检测周期较长,通常需要2周以上。该方法不仅对DNA的纯度和浓度要求较高(根据不同的作物DNA的需求量在20~100 μg),而且对人员操作的技术要求比较严格,期间很多中间环节(如,DNA提取质量不佳、酶切效果不好、转膜不充分、杂交条件或漂洗不能使阳性结果与背景产生明显反差等)都可能导致无法得到准确的检测结果,且重复性差,使原本较长的检测周期二次延长,且容易在内切酶的选择上受到限制,因此,自2014年来转基因作物中外源基因的拷贝数鉴定方法,逐渐被数字PCR等新兴技术所取代。
数字PCR技术已经在转基因水稻、转基因玉米和转基因小麦等转基因作物的外源基因拷贝数鉴定中广泛应用,且在基因表达研究、癌症标志物稀有突变检测和转基因成分鉴定等诸多领域显示出广阔的应用前景。本研究基于微滴数字PCR技术,建立了转基因大豆‘GE-J12’中外源基因拷贝数的分析方法。该方法操作步骤简便,仅需DNA提取、PCR扩增、微滴生成与读取等常规步骤,即可在6~8 h完成对转基因作物外源基因拷贝数的鉴定,不同试验人员间可稳定重复。同时该方法对DNA需求量最低只需200 ng,远远低于Southern杂交所需DNA的量,且结果与Southern杂交一致。因此本方法具有快速、简便、高效、不依赖任何标准品、所需样本量少、不受内切酶位点选择限制等优点。
在进行转基因作物微滴数字PCR拷贝数鉴定之前,通常需在该作物外源插入序列的两侧基因组DNA序列上和外源插入序列的5′或3′设计2对定性PCR引物,且2对引物共用同一上游或下游引物,应用这2对引物的扩增结果对植株进行纯合性鉴定。而本研究在建立转基因大豆‘GE-J12’中外源基因拷贝数分析方法的同时,应用3′端转化体特异性序列可监测植株纯合性,无需进行前期的定性PCR引物设计及扩增,简化了试验步骤,且保证了试验结果的准确性。该方法不仅仅局限于外源目的基因G
2-EPSPS
和GAT
,同时也可应用于该作物基因型整合鉴定中所含有的CaMV
35S
启动子、CaMV
35S
终止子和NOS
终止子等调控元件的拷贝数分析,从而实现通过一步反应对该品种T-DNA外源插入片段不同部位多种整合基因型拷贝数的全面分析。后续试验可对多重微滴数字PCR检测体系的建立进行摸索,进一步提高试验准确性、节约试剂成本、提高试验效率,同时对比NGS技术分析外源插入序列拷贝数,为传统方法无法解析的复杂整合体的分子特征奠定试验基础。4 结 论
基于微滴数字PCR技术,以转基因大豆‘GE-J12’转化体的外源插入目的基因G2-EPSPS
和GAT
的序列、3′转化体特异性序列为靶序列,设计了3对引物及探针,以大豆Lectin
内标基因为参照,建立了微滴数字PCR拷贝数检测方法。应用该方法,鉴定G2-EPSPS
基因、GAT
基因和3′转化体特异性序列的拷贝数均值分别为0.99、1.01和1.00,均接近于1个拷贝,结果表明,转基因大豆‘GE-J12’的外源基因在大豆基因组上为单拷贝插入,且为纯合植株。该方法特异性强、准确性高、重复性稳定,操作简便、快速,在6~8 h完成对转基因作物外源基因拷贝数的鉴定。