基于机器学习的β-内酰胺酶注释预测和分析平台
2022-01-05蓝小斌陈嘉豪管军霖
侯 镱, 蓝小斌, 陈嘉豪, 管军霖
(桂林电子科技大学 计算机与信息安全学院,广西 桂林 541004)
β-内酰胺类抗生素是临床实践中应用最广泛的抗菌药物之一,主要用于治疗病原体引起的感染。但由于这一类抗菌药物的滥用和误用,病原菌对相应药物产生了耐药性,严重影响了药物的治疗效果并且限制了临床医生的用药选择[1]。目前研究发现,绝大多数病原菌是通过分泌β-内酰胺酶水解抗生素药物中的β-内酰胺基环,进而破坏蛋白质分子结构,导致药物活性降低,从而使细菌具有抗药性[2]。因此,进一步研究β-内酰胺酶序列和蛋白质结构的生物学特性,完善其分类,有利于深入分析相关蛋白质的耐药机制。
近年来,β-内酰胺酶蛋白质的研究主要有以下3个特点:
1)蛋白质数据的迅速增加,截至目前,NCBI数据库(常用生物学蛋白质数据库)中β-内酰胺酶数据量由2017年的1 022 470条扩展至2020年的3 477 250条;
2)β-内酰胺酶蛋白质家族分类日渐繁多,且不同家族的蛋白质特性之间差异较大;
3)病原体在选择压力下变迁,导致β-内酰胺酶变异体数目的增加,并进一步增加蛋白质序列和注释信息之间的差距[3-4]。
以上变化使得选择生物实验作为研究β-内酰胺酶的科研手段变得耗时且费力,随着机器学习方法在生物领域的迅速发展,发现机器学习方法不仅可对海量数据进行高效分析和处理,且所拥有的准确率高、性能稳定等优势可有效弥补生物实验手段验证过程中存在的不足,加速探索进程。
目前,提供针对β-内酰胺酶的注释和分析的数据库主要有BLAD[5]、BLDB[6]、CBMAR[7]、AIBLDB[8]和LaCED[9]。这些平台的功能和数据集的统计情况如表1所示。已有的β-内酰胺酶数据库不同程度上实现了对蛋白质注释信息的收集和展示,并在系统分析上使用BLAST序列比对,系统亲缘生成树构建和序列家族指纹方法对蛋白质的注释信息作进一步补充,但它们仅收集单个家族或者部分蛋白的数据和注释,仅实现了β-内酰胺酶的数据收集、查询、注释和展示,并未提供对潜在蛋白质的分析和预测识别,功能模块之间没有相互关联,不能满足用户预测分析的需求[10-11]。基于以上不足,开发了BLHub平台,用于对潜在蛋白进行预测和分析。首先通过系统查阅β-内酰胺酶相关文献,收集蛋白质数据,并且根据关键字在UniProt或者NCBI公共数据库进行检索和下载[12-13];接着对从5个相关文献和公共数据库收集的蛋白质数据进行整合,剔除和纠正错误序列;根据蛋白质索引收集相关的注释信息,包括β-内酰胺酶蛋白质的序列、功能和结构等。为了进一步探索和分析蛋白质的结构和功能,BLHub采用支持向量机对已经收集的β-内酰胺酶序列进行机器学习建模,用于对潜在蛋白质的预测和分析[14]。BLHub平台整合BLAST多序列比对分析和Phylogenetic系统亲缘分析,用户可以快速分析并准确定位潜在蛋白质同源物,把BLAST比对和Phylogenetic亲缘分析结果通过重定向技术返回BLHub的数据库注释信息页面进行深入分析。

表1 BLHub平台与5个β-内酰胺酶蛋白质数据库之间的比较
1 材料与方法
BLHub平台主要包括以下3个部分:蛋白质数据收集和管理、蛋白质信息注释和BLHub平台的功能(如图1所示)。
1.1 蛋白质收集和管理
系统地查阅现有的β-内酰胺酶蛋白质的文献资料和公共数据库(UniProt和NCBI),从已经公布的β-内酰胺酶文献中收集11 022条蛋白质,从公共数据库中收集经过审查的β-内酰胺酶蛋白质879条。由于数据库不提供数据下载功能,本实验使用爬虫等技术手段对数据库中数据进行收集,其中BLAD、BLDB、CBMAR、AIBLDB和LaCED数据库中蛋白质数量分别为1 894、4 405、2 160、1 174和1 389条,收集到的数据与已发表文献中蛋白质数量存在一定差异。此外BLAD和AIBLDB数据库由于客观因素暂时不能访问,但是上述2个数据库收录的2篇相关文章中提及β-内酰胺酶的研究方法优先于BLHub,所以暂时保留与BLAD和AIBLDB数据库比对部分。在数据汇总后,为了保证数据的准确性,需要进行预处理,首先根据Ambler分类法将蛋白质分为A、B、C和D类,并且使用CD-HIT工具对数据去冗余(阈值设置为1),即去除百分百相似序列,保持蛋白质独一无二[15],4种不同类型的β-内酰胺酶分别是1 756、913、1 443和976条;接着剔除不精确的蛋白质(蛋白质注释信息标注为“partial”的序列,表示该序列不完全是β-内酰胺酶),得到A、B、C和D类蛋白质数量是1 709、908、1 410和903条;最后将蛋白质序列索引号与NCBI或UniProt进行比对,手动检查更新每条蛋白质信息,得到1 339条A类蛋白质,832条B类蛋白质,1 381条C类蛋白质和854条D类蛋白质。BLHub的数据库最终收录了4 406条蛋白质(来源602个物种),分别是1 339条A类蛋白(来源219类物种)、832条B类蛋白(来源236类物种)、1 381条C类蛋白质(来源81类物种)和854条D类蛋白质(来源66类物种),β-内酰胺酶数量和物种统计分布如图2所示。
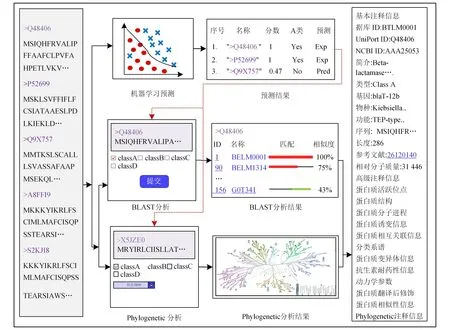
图1 BLHub平台的主要框架

图2 蛋白质种类和物种数量条形统计
1.2 蛋白质注释信息
BLHub收集的蛋白质基本信息分别包括BLHub ID、UniProt ID、NCBI ID、蛋白质概要、蛋白质类型、基因信息、物种来源、蛋白质功能、蛋白质序列、蛋白质序列长度、蛋白质来源的PubMed号以及蛋白质相对分子质量。除此之外,BLHub针对性地整合了更为复杂的注释信息,包括来自于PDB数据库的蛋白质三维结构信息、来自于STRING[16]和MINT数据库的蛋白质相互作用、UniProt数据库的蛋白质活跃位点、蛋白质分子进程、序列诱变信息、蛋白质分类谱、变异体信息、抗生素耐药性信息、动力学参数以及蛋白质翻译后修饰信息[17]。对于每条蛋白质,数据库针对已知蛋白质提供的序列信息进行预先计算和预测注释分析:1)预先计算使用BLAST工具(version 2.8.1+)搜索已知蛋白质以及进一步计算序列之间相似性,利用BlasterJS实现可视化展示[18];2)预测注释分析使用MAFFT工具(version 7.271)对已知蛋白质进行亲缘性分析和系统发育演化的计算,通过Fast-Tree(version 2.1.8)构建发育生成树,并利用jsPhyloSVG进行可视化分析[19]。
1.3 数据库的基本功能
BLHub平台包括以下6个功能:1)使用Echart可视化软件对蛋白质的种类、蛋白质来源以及蛋白质基因类型的数量进行统计和可视化展示;2)对4种不同类型蛋白质的基本信息进行浏览和数据的下载;3)使用Database ID、UniProt ID和NCBI ID关键字进行蛋白质的精确搜索,或者使用基因名称和蛋白质来源等关键字进行模糊查询;4)通过支持向量机模型对用户提交的序列进行预测;5)分别使用BLAST比对和系统亲缘分析,对潜在蛋白质进行序列相似性和蛋白质进化演化分析。
1.4 平台实现技术
BLHub平台使用JAVA(https://www.java.com/)进行服务器开发,Structs2(https://structs.apache.org/)架构用于实现BLHub的数据库分析平台的业务逻辑,Hibernate(https://hibernate.org/)用于进行模型层的蛋白质数据的传输。视图层由JSP、CSS和jQuery(https://jquery.com/)实现。前端页面中蛋白质数据加载和信息展示使用Bootstrap框架(http://bootstrapdocs.com/)。MySQL数据库作为蛋白质(http://www.mysql.com/)和注释信息相关数据的存储介质。通过在队列服务中使用Perl CGI(https://metacpan.org/)工具构建自动化流程分析过程,并使用Gearman框架(http://gearman.org/)对子任务处理系统进行工作调度,将最终结果输出到Web前端界面。
2 BLHub平台数据库模块
BLHub包括平台基本模块、预测、关系分析、帮助和联系5个标签页。所有功能模块都可以在BLHub(如图3所示)中独立运行或者进行模块之间互联互通,BLHub平台的各种功能模块在“帮助”界面中有详细说明。

图3 BLHub平台中预测分析模块和数据库模块之间的交互
2.1 浏览
该页面中的β-内酰胺酶按分类进行组织,每条蛋白质都展示其 BLHub ID、基因名称、简短描述、种类、UniProt ID、NCBI ID和PubMed ID等基本信息。浏览页面的表中包含搜索功能,可快速查找指定的蛋白质。通过点击唯一的BLHub ID链接,搜索结果被定向到相应蛋白质的信息页面,以进行蛋白质的注释和分析,或者用户可以单击UniProt ID、NCBI ID或PubMed ID,通过链接跳转到相应的外部资源。
2.2 搜索
与“浏览”页面的功能相比,此页面为用户提供更高级的搜索选项。高级搜索功能可通过蛋白质的基因名称或物种来源等关键字进行模糊查询,也可以根据BLHub ID、UniProt ID或NCBI ID等关键字进行精确查询。
2.3 数据展示和下载
该页面包含蛋白质数据统计结果的可视化功能,统计数据包括蛋白质的种类、蛋白质基因名称和蛋白质来源。通过Echart工具对4种不同种类β-内酰胺酶的数据进行统计和可视化展示。此外,本平台支持SQL格式数据库文件、Fasta格式序列文件和Aln格式比对文件的下载。
2.4 蛋白质数据展示模块
该页面为每条蛋白质提供了详细的注释信息,包括蛋白质基本注释信息、高级注释信息以及其相关联的信息。基本注释信息包括BLHub ID、UniProt ID、NCBI Protein ID、基因名称、简短描述、蛋白质类型、基因注释信息、功能、序列、序列长度和PubMed ID,高级注释则包含蛋白质三维结构信息、蛋白质活跃位点、蛋白质结构、蛋白质分子进程、序列诱变信息、蛋白质相互作用、蛋白质系谱、变异体信息、抗生素耐药性信息、动力学参数、蛋白质翻译后修饰和系统发育注释信息,蛋白质注释信息在1.2节中有详细描述[20-21]。
3 机器学习预测模块
3.1 数据集处理
为了从功能信息未知的序列中识别出潜在β-内酰胺酶,BLHub提供的基于支持向量机预测模型对用户提交的潜在序列进行识别。建模过程中使用的β-内酰胺酶共4 406条,经过CD-HIT(阈值设置为0.5)去冗余后,保留184条作为正样本,其中A、B、C和D类的数量分别为61、86、11和26条;负样本为从UniProt数据库中随机挑选的1 000条非β-内酰胺酶,经过去冗余后,保留478条蛋白质。收集的β-内酰胺酶蛋白质数据经过预处理后结果如表2所示。
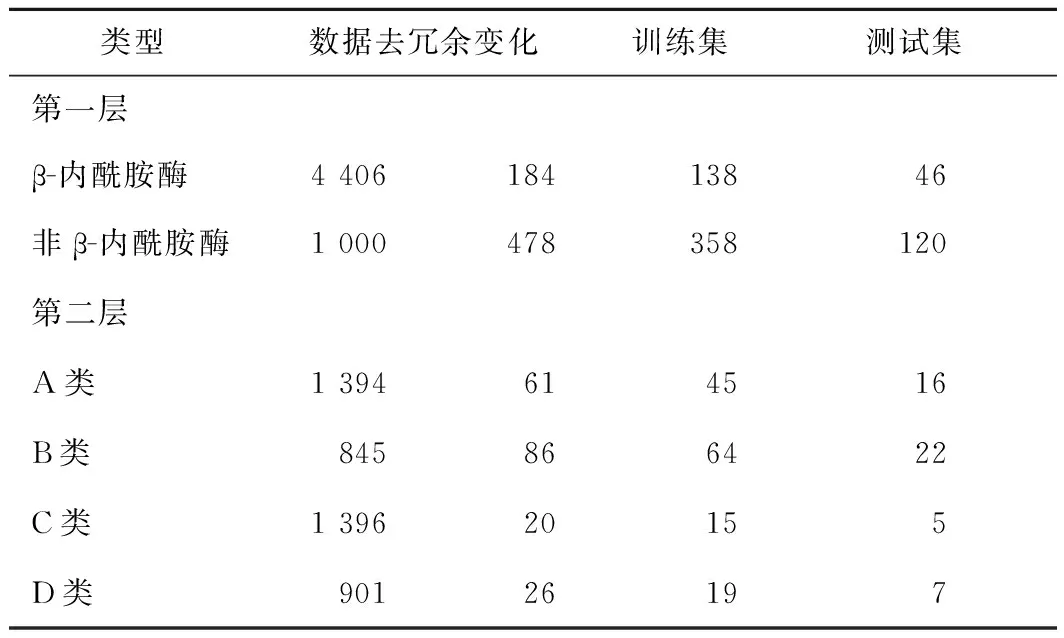
表2 对机器学习建模的数据集进行预处理后的结果
3.2 预测模型建立
目前,很多机器学习预测工具可以对生物蛋白质数据进行建模、预测和分析,如K-近邻、神经网络、逻辑回归和支持向量机,其中支持向量机(support vector machine,简称SVM)是应用最广泛的机器学习算法,在解决分类、回归以及评估等问题时具有较强的学习能力和泛化能力。近年来,SVM在蛋白质分子预测分析领域得到了广泛应用,如用于蛋白质位点预测和蛋白质结构预测等方面[22-23]。SVM主要通过数据集特征提取、建模,找到一个最优的分隔面,并将其作为分割边界[24]。
预测模型构建过程如下:首先构建第1层SVM模型,预测、识别潜在序列是否为β-内酰胺酶,对于预测为正样本的序列继续分析,通过第2层SVM模型进一步预测潜在蛋白质的种类。因此,为了处理多分类的问题,本算法使用One-vs-Rest多分类方法,将某种类别的蛋白质视为正样本、其余类别的蛋白质视为负样本,并进行数据集的分类。
3.3 预测模型性能评估
在模型训练和测试的过程中,用5折交叉验证训练支持向量机预测模型,训练集被随机划分为5个大小大致相同、互不相交的子集,选择4个作为训练集,剩下1个作为测试集[25]。为了定量评估独立测试期间生成模型的预测性能,使用敏感度(SN)、特异性(SP)、准确性(ACC)、F值(F-value)和马修斯相关系数(MCC)对机器学习性能进行评估。
3.4 结果分析
交叉验证和独立测试集的结果如表3、4所示。从表3、4可看出,BLHub对潜在β-内酰胺酶进行分类,其中A、B、C、D类预测模型对应的MCC值分别为0.76、0.586、0.515、0.914,A类模型和D类模型预测性能较好;GTPC、PDT特征编码方式可以有效提取A类蛋白质序列的信息;RPM-PSSM、CTDT编码方式可以有效提取B类蛋白质的有效信息;CKSAAGP、DR、PseKRAAC、RPM-PSSM特征编码方式可以有效提取D类蛋白质的有效信息。
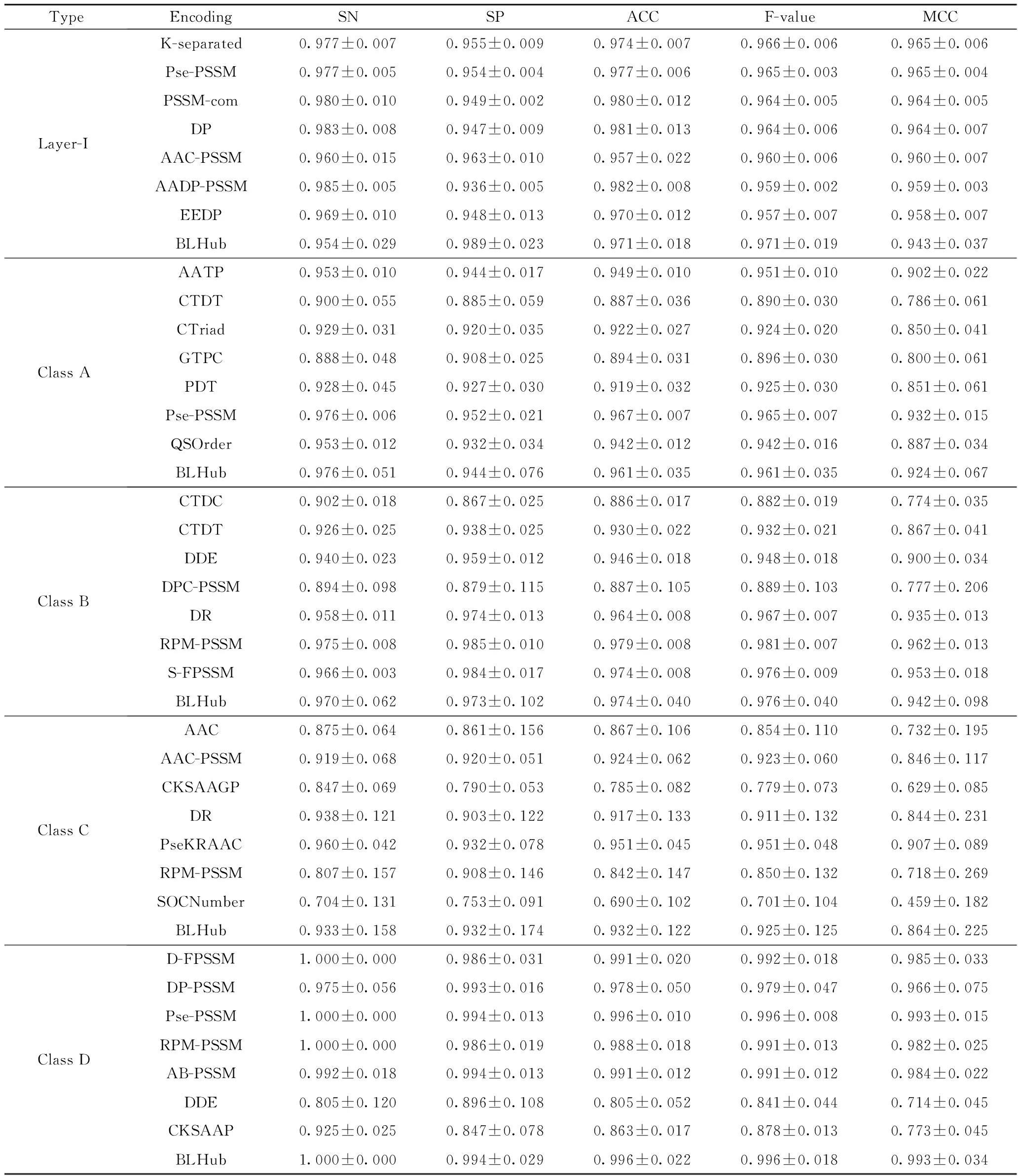
表3 5折交叉验证评估支持向量机预测模型的性能
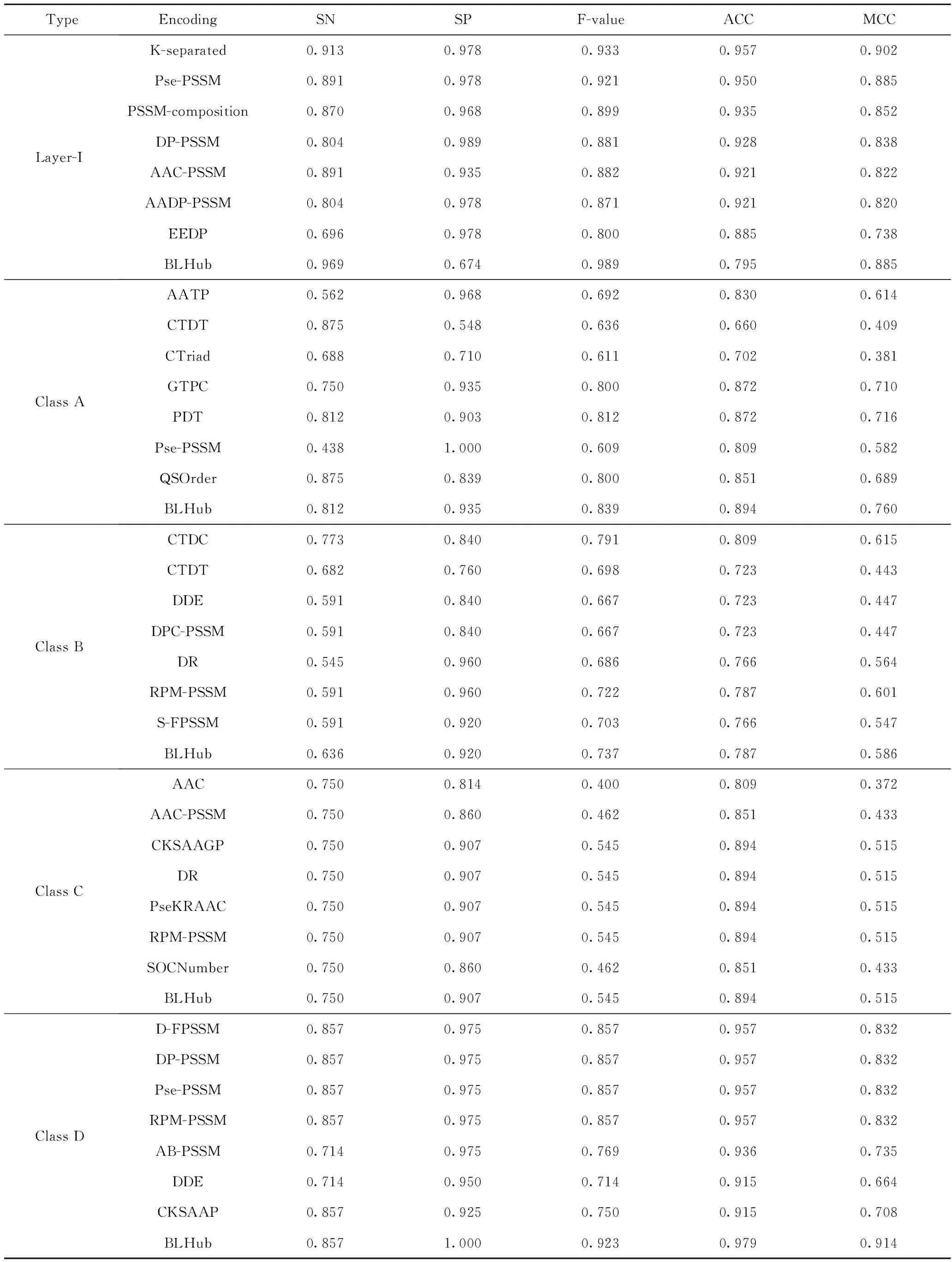
表4 独立测试集来评估支持向量机预测模型的性能
4 关系模型分析模块
本研究分析了潜在的序列和已知的β-内酰胺酶之间的序列关系,用于启发、推断、预测蛋白质的特征。然而由于机器学习模型不能显著地表示潜在蛋白质和已知蛋白质的同源关系,所以通过进一步集成2个关系分析模块分析潜在蛋白质遗传和系统的亲缘关系。虽然BLHub中的所有功能模块都是作为独立服务项来设计和实现的,但是为了方便分析和使用,模块之间数据可以自动传递,形成如下分析流程:
1)机器学习模型的预测结果可以直接指定为2个关系模型的输入,用来进一步分析潜在蛋白质与已知蛋白质之间的相似性和亲缘关系。
2)预测模型关系中的分析结果通过链接重定向对应的数据库注释信息界面:标记为“Exp”的蛋白质,提供可跳到相应详细信息页面的链接供用户查阅;而标记为“Pred”的表示预测出来的β-内酰胺酶蛋白质,通过重定向来分析潜在蛋白质和已知蛋白质之间的关系。
4.1 序列相似性分析
通过序列比对计算结果寻找潜在蛋白质与已知蛋白质的关系,并通过BlasterJS工具将比对结果进行可视化展示。可视化展示主要分为3部分:第1部分中的下拉框可以查看任意的BLHub ID,单击链接可以跳转到对应的蛋白质注释信息界面;第3部分为潜在蛋白质和已知蛋白质序列之间氨基酸的比对过程,用于展示序列之间的突变和相似序列片段。
4.2 系统亲缘分析
对于潜在的β-内酰胺酶蛋白质,在BLHub平台中,用户先选择特定种类的β-内酰胺酶数据集,将任务序列提交到后台服务器后进行计算,并分析潜在蛋白质和已知蛋白质之间的序列同源性。系统亲缘分析与多序列相似性比对过程存在明显区别:在构建过程中需要考虑时间的变化对蛋白质序列和功能造成的影响。例如2条不同的蛋白质片段AEFT和AEFF,根据距离法判断是高度相似的片段,但若考虑时间因素,则造成蛋白质功能的差异就可能不同。例如AEFT片段通过10次变异进化,然而AEFF片段通过5次变异进化,从系统亲缘角度分析,2条蛋白质无亲缘性,但是从多序列比对的角度来分析,2条蛋白存在相似性,因此系统亲缘分析是对多序列比对分析的重要补充。在系统亲缘结果中描述潜在蛋白质和已知蛋白质之间的关系,并依靠已知蛋白质链接重定向到相应的注释信息界面。
5 结束语
通过BLHub平台对潜在蛋白质功能和性质进行了深入分析和研究。首先通过公共数据库和参考文献收集β-内酰胺酶蛋白质序列,并从UniProt等公共数据库中收集抗生素耐药性、活跃位点、蛋白质突变信息以及蛋白质翻译后修饰等注释信息,并将收集的数据存入MySQL数据库;然后用机器学习方法对潜在蛋白质的种类进行预测,通过BLAST序列比对工具和Phylogenetic亲缘性分析工具对机器学习预测的结果进行序列同源性和系统亲缘性分析,找出与潜在蛋白质最相似的β-内酰胺酶;最后通过重定向的方式返回到指定的蛋白质注释信息页面,根据已知蛋白质的性质和功能推测潜在蛋白质的性质和功能。
BLHub意在帮助和引导相关学者进行蛋白质分析和进行下一步的实验设计和验证,目前该平台已经收集了完善的蛋白质相关注释信息,但缺少相应基因的注释信息。在未来的工作中,构建一个同时包含基因信息和蛋白质信息的数据库分析平台,将有助于研究者进一步了解β-内酰胺酶的功能和特征。
