鞘氨醇激酶1抑制剂的研究进展
2021-12-31汪小涧
杨 倩,汪小涧
(1国家知识产权局专利局专利审查协作北京中心,北京100160;2中国医学科学院、北京协和医学院药物研究所,天然药物活性物质与功能国家重点实验室,北京100050)
鞘脂(sphingolipids)是生物膜的重要组成部分,具有两性结构,两端分别是长链脂肪酸和极性醇[1]。在生物体内,鞘脂可通过溶酶体中的神经磷脂酶催化水解生成神经酰胺(ceramide,Cer),然后被神经酰胺水解酶水解生成鞘氨醇(sphingosine,Sph),再 通过 鞘氨 醇激 酶(sphingosine kinase,SphK)磷酸化得到鞘氨醇-1-磷酸(sphingosine 1-phosphate,S1P)。S1P也可以通过鞘氨醇-1-磷酸磷酸酶转化为Sph,进而在神经酰胺合成酶的作用下重新生成Cer。Cer-Sph-S1P 之间可逆的动态平衡被称为鞘脂-变阻器(sphingolipid-rheostat)。其中,SphK作为催化Sph生成S1P过程中的限速酶,在调控该平衡时起重要作用[2]。研究表明,SphK 抑制剂可以使Cer/Sph 的比例升高以及S1P 含量降低,从而促进肿瘤细胞凋亡,具有良好的药物开发前景[3-6]。
1 SphK1的结构生物学
SphK 分为SphK1 和SphK2 两种亚型,序列高度同源,但是在分布和功能上有一定差异。从器官和组织分布来看,SphK1 在脾和肺中含量较高,SphK2 在心脏和肝中含量较高;从细胞内分布来看,SphK1 主要存在于细胞质,SphK2 主要存在于细胞核。从对Cer-Sph-S1P 平衡的调控作用来看,下调SphK1 的表达时平衡向Cer 方向移动,而下调SphK2 的表达时则会促使平衡向S1P 方向移动[6]。其中,SphK1参与肿瘤发生发展过程的调控作用受到较多关注。
SphK1 的激活会引发一系列信号通路级联传递,涉及PI3K/AKT/GSK3 通路和S1P/S1PR3/Notch等通路[7-8]。SphK1 具有致癌作用,在肺癌、乳腺癌、胃癌、结肠癌等多种肿瘤细胞中SphK1 的表达和mRNA 水平明显高于正常细胞[9]。Olivera 等[10]发现,NIH/3T3 成纤维细胞中SphK1 活性提高后S1P 含量随之升高,使得细胞增殖速度明显加快。同时,SphK1 活性提高还可以使NIH/3T3 成纤维细胞和HEK293 细胞在无血清培养或高浓度神经酰胺条件下免于凋亡,具备了类似肿瘤细胞的增殖能力。Xia 等[11]将SphK1 高表达的SK-3T3 细胞移植到小鼠体内,四周后所有的小鼠都发生了癌变。SphK1 也具有促进肿瘤组织血管生成的作用[12]。Nava 等[13]发现注射高表达SphK1 的MCF-7 细胞的裸鼠肿瘤组织周围的血管密度增加。SphK1 活性也与肿瘤细胞对化疗或放疗的敏感性相关。Min等[14]发现加入SphK1 抑制剂后黏菌细胞对顺铂的敏感性提高。Sauer等[15]发现抑制SphK1的活性可以降低前列腺癌细胞对多西他赛的耐药性。Bonhoure 等[16]发现对伊马替尼有耐药性的LAMA84-s细胞中SphK1 高表达,当使用SphK1 抑制剂F-12509a 抑制SphK1 的活性或者用siRNA 沉默SphK1 的表达时,耐药性得到显著改善。 Nava等[17]发现对放疗不敏感的前列腺癌细胞LNCaP 中SphK1活性较正常细胞更高,抑制SphK1的活性后伽马射线诱导LNCaP凋亡的效果显著增强。
近期研究结果显示:SphK1 和S1PR3 受体与GC 细胞的趋化性和调节密切相关[18]。在血液中存在的鞘脂溶血磷脂酸处理的MKN1 GC细胞中观察到SphK1 mRNA 和蛋白水平的增加,而SphK2的表达不受影响。SphK1 和S1PR3 受体介导LPA 和EGF刺激的MKN1细胞迁移和侵袭,但LPA调节的新生血管形成因子(包括白细胞介素)的表达不受该激酶影响。
目前,人源SphK1晶体结构以及SphK1与底物鞘氨醇的共结晶结构已经被报道[19]。SphK1 由N-末端和C-末端两个结构域组成,包含9个α 螺旋和17 个β 折叠结构。在SphK1 的C 端有一个J 型口袋,是底物鞘氨醇的结合部位。鞘氨醇结构中的脂肪链与J 型口袋的多个氨基酸残基之间形成疏水作用,2-氨基-1,3 二醇末端与Asp81,Asp178 和Ser168 形成氢键作用。SphK1 的ATP 结合域位于N-末端和C-末端间的裂缝区域,ATP 的碱基与Glu55,Arg56 和Asn22 之间形成π-π堆积作用,磷酸基团与Asp341,Glu343,Arg185和Arg191形成氢键和静电盐桥作用。鞘氨醇和ATP 在SphK1 的结合位点在空间上较为接近。近年来,研究者们通过对底物鞘氨醇进行结构改造或高通量筛选新结构分子等多种手段进行SphK1 抑制剂的研究。本文根据抑制剂的结构特征和来源进行分类,并对各类抑制剂的构效关系进行综述。
2 SphK1抑制剂的结构类型和构效关系
2.1 鞘氨醇(Sph)类似物
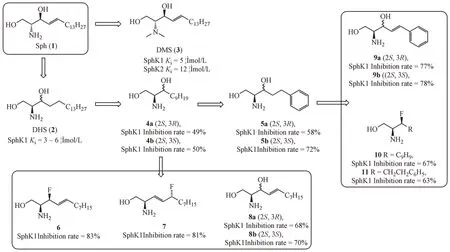
Sph(1)是SphK 的天然底物。从Sph 出发,将结构中的不饱和烯链替换为饱和脂肪链得到DHS(2)对SphK1有一定的抑制活性[20]。将Sph的氨基进行二甲基化后得到DMS(3),对SphK1 和SphK2均有抑制活性[21]。De Jonghe 等[22]进一步围绕Sph开展结构改造研究,包括缩短脂肪链的长度(4),在短链中引入苯环(5,9 ~11),去除或改变双键的位置(7),将羟基替换为氟等(6,8)。构效关系研究表明,缩短脂肪链长度不会明显影响SphK1 抑制活性,引入苯环、氟原子取代羟基、改变双键的位置等均能提高抑制活性,羟基或氟取代的碳原子构型对活性没有太大影响。
2.2 环状氨基醇类
FTY-720(12)是诺华制药研发的S1P 受体激动剂,于2010年获批上市,用于多发硬化症的治疗。研究发现FTY-720 也是SphK1 的底物竞争性抑制剂,可以诱导SphK1 蛋白降解。Xiang 等[23]将氨基丙二醇片段替换为五元环状氨基醇片段,并通过酰胺键与苯环相连得到化合物13,对SphK1具有较强抑制作用,但是水溶性较差。首先,尝试在烷基侧链中引入五元芳杂环得到化合物14,虽然提高了水溶性,但是SphK1 的抑制活性显著降低。随后在化合物14的苯环和酰胺氮之间引入亚甲基获得化合物15的抑制活性显著提高。继续优化烷基侧链,改变烷基侧链的长度,将链状烷烃替换为环烷烃或苯环等最终得到化合物16的活性和水溶性都有提高[24]。
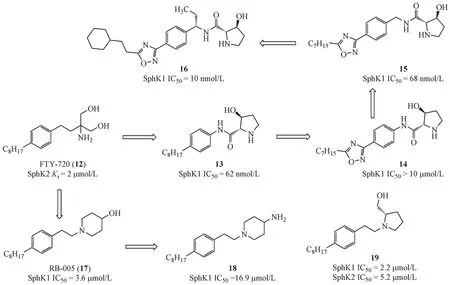
Baek 等[25]将FTY-720 的氨基丙二醇片段替换为六元环状氨基醇片段,经过筛选发现4-羟基哌啶的活性最强,得到RB-005(17)。研究表明,RB-005 除了能够抑制SphK1 的活性外,还能促进SphK1蛋白发生降解。构效关系研究显示,哌啶环的羟基作为氢键供体是化合物具备SphK1 抑制活性的必须基团,将羟基替换为氨基(18)或将哌啶六元环换为五元环(19)都能保持活性,但是将哌啶的4-位羟基甲基化或氧化为羰基,缺失氢键供体则活性全部丧失。
辉瑞公司的研究人员在高通量筛选的基础上结合结构优化获得PF-543(20),对SphK1 的抑制活性Ki达到3.6 nmol/L,对SphK2 的抑制活性仅为360 nmol/L,是目前报道的活性最强的SphK1 选择性抑制剂。分析PF-543 与SphK1 的共结晶复合物可以看出,PF-543 采用一种弯曲构象与SphK1 结合,其末端的苯环伸展到由苯丙氨酸,亮氨酸等组成的疏水口袋中,环状氨基醇极性片段靠近ATP结合位点,氮原子和羟基与影响底物识别的关键氨基酸残基Asp264 的侧链形成氢键作用(图1)。PF-543 产生SphK1/SphK2 选择性抑制作用的效果也可以通过分子结合模式得到印证。PF-543 结构末端苯环可以与SphK1 的芳香氨基酸残基Phe374之间形成π-π堆积作用,而SphK2该在相应位置的氨基酸残基是Cys374,无法与苯环形成相互作用,导致结合力显著下降。除了对SphK1 抑制作用的选择性外,PF-543 也可以作为底物被SphK1 磷酸化,但是磷酸化的PF-543 并不会与S1P 受体结合,即不会产生与S1P 类似的生物活性[26-27]。Zhang等[28]近期报道,PF-543 通过降低S1P 水平可显著减少镰刀状红细胞的生成,提示PF-543 可以被应用于治疗镰刀细胞贫血症。对比在先报道的鞘氨醇类SphK1 抑制剂,PF-543 的亲脂区域的磺酰苯环以及甲苯结构与SphK1 的亲脂区域有更强的疏水结合作用,而亲水作用的区域则可以与天冬氨酸264形成静电盐桥的作用力,提高了其对蛋白的结合活性。
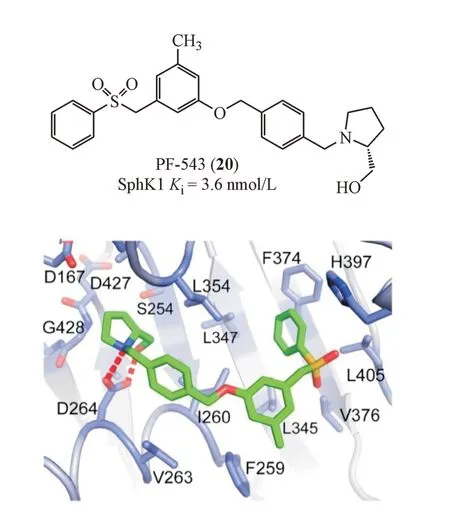
图1 PF-543(20)与鞘氨醇激酶1(SphK1)的结合模式
2.3 末端为脒基或胍基极性结构
除了环状氨基醇结构,对氨基醇的改造还包括将其替换为脒基或胍基。Foss 等[29]报道了一类具有脒基末端的鞘氨醇激酶抑制剂,在S1P激动剂研究中获得活性化合物VPC4512(21),进一步探索极性末端的修饰方式获得化合物22 具有SphK抑制作用。将脒基替换为极性的羧基(23)和酰胺(24)后抑制活性完全丧失。分析原因,脒基质子化带有正电荷,类似于鞘氨醇的胺基,可以与SphK1 的Asp177 形成盐桥作用,对抑制活性至关重要,羧基在生理条件下带负电荷,酰胺为中性,均不能形成盐桥作用,因而活性丧失。进一步对手性中心进行改造,将甲基替换为环丙基消除手性同时增大立体体积,得到化合物25,与化合物24相比对SphK1 的选择性增强1 倍。将化合物25 的烷基链替换为不同长度的脂肪链,结果显示12 个碳原子的长度为最佳(26),调换酰胺侧链方向(27)在保持SphK1 抑制活性的同时SphK2 抑制活性降低为原来的1/10,显示出更优的SphK1选择抑制作用。第二轮结构改造中,将化合物26 的环丙基替换为环丁基(28),或是骨架跃迁为2-脒基四氢吡咯结构得到化合物29 和VPC96091(30),然而化合物28和29的活性都显著下降,仅化合物30活性保持,表明脒基与酰胺键的二面角对于活性有重要影响。进一步将化合物30的脂肪链替换为苯环、环戊烷、环己烷、金刚烷等体积更大的侧链考察对活性的影响,引入醚键调整化合物的clogP,最终得到的化合物31 对SphK1 的抑制活性有所增强。但是,化合物31 结构的自由度较大可能影响结合力,考虑适当增强刚性,保留脒基极性末端和环己烷脂肪侧链获得化合物32,对于SphK1 的抑制活性进一步提高[30]。
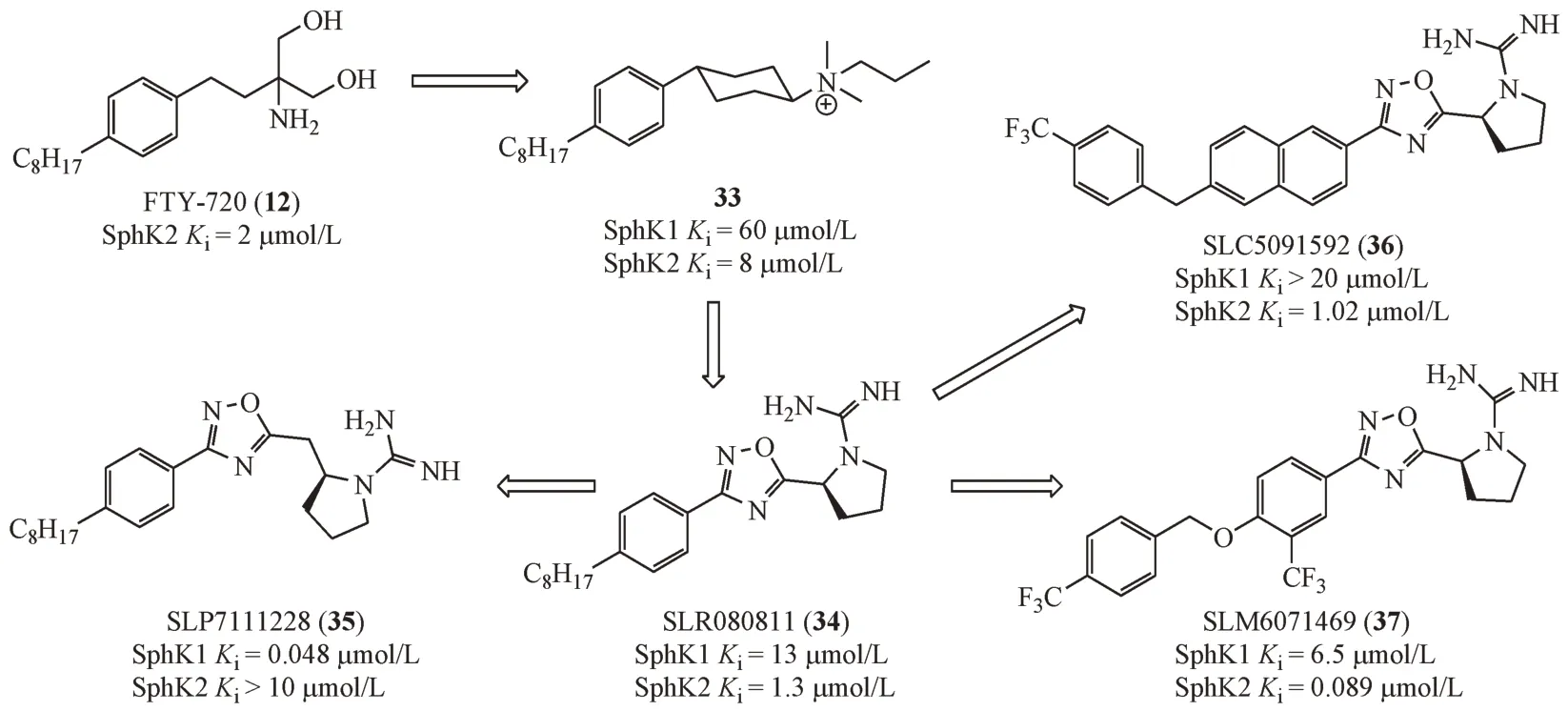
在FTY-720(12)的基础上,将其氨基丙二醇片段替换为伯胺、仲胺、叔胺和季铵盐结构的衍生物,其中化合物33 具有中等水平的SphK1/2 抑制活性和微弱的SphK2 选择性。将季铵盐结构替换为胍基,在生理条件下,胍基与脒基性质相似,其质子化后带有正电荷,同样可与Asp 残基产生氢键、盐桥等相互作用。进一步将环己烷结构替换为刚性更强的芳杂环结构,增强配体与蛋白之间的结合力,得到SLR080811(34)对SphK1 和SphK2均有一定的的抑制活性。值得注意的是,在噁二唑环和四氢吡咯环中间引入一个亚甲基后可增强对SphK1 的选择性(35)[31]。将化合物34 的苯环替换为萘环骨架,在萘环上引入不同的醚键并进行取代基的修饰,结果显示对位三氟甲基取代的苄氧基化合物SLC5091592(36)对SphK2 的选择性显著提高[32]。进一步修饰脂肪区的体积和亲脂性得到SLM6071469(37)还可以进一步提高选择性[33]。

2.4 通过高通量筛选获得的五元芳杂环类
2003年,French等[34]通过高通量筛选发现4个新结构类型的SphK 抑制剂,分别命名为SKI-I(38)、SKI-II(39)、SKI-III(40)和SKI-IV(41),这些化合物对于多种激酶有抑制作用,其中SKI-II 对SphK1 具有选择性抑制作用,毒性较小,表现出较高的口服生物利用度和适宜的半衰期(t1/2=15 h)。除了抑制SphK1 活性外,SKI-II 可以激活SphK1 的泛素-蛋白酶降解途径,促进SphK1 蛋白在体内被降解。正常生理条件下,SKI-II 可显著缩短SphK1蛋白的半衰期至0.8 h[35],并且可以促进SphK1 被溶酶体的Cathepsin B 迅速降解。各项研究结果均提示SKI-II是一个优质的先导化合物[36]。

Aurelio 等[37]在SKI-II 结构基础上将噻唑环电子等排替换为噁二唑环后得到化合物42,对SphK1/2 的抑制活性略微提高,抑制肿瘤细胞PC3增殖活性提高达10倍。进一步将氨基连接链省略得到化合物43,对SphK1/2 的抑制活性没有变化,但抑制肿瘤细胞PC3 增殖的能力却丧失。 在SphK1蛋白降解实验中,含噁二唑环结构的化合物42对SphK1的抑制能力比SKI-II强,却没有像SKIII 一 样 诱 导SphK1 的 降 解,而 化 合 物44、45 对SphK1 的抑制能力虽然比SKI-II 弱,却能够有效诱导SphK1的降解,表明诱导SphK1降解的能力与抑制SphK1 活性并非直接相关。鉴于SphK1 蛋白降解与变构位点有关,因此推测化合物42 诱导SphK1 降解能力丧失可能是因为SKI-II 噻唑环换成噁二唑环后与变构位点的亲和力下降所致。化合物SKI-II 和化合物42 均具有抑制二氢神经酰胺脱氢酶(Des 1)的活性,其结构中的对氨基苯酚片段可被Des 1 氧化为亚胺醌结构,进而与Des 1 酶发生亲核加成而共价结合,从而不可逆的抑制Des 1。SKI-II 衍生物46 中不含对氨基苯酚片段,则没有Des 1 的抑制活性。虽然化合物46 对SphK1 和SphK2 具有一定的抑制活性,但抑制肿瘤细胞PC3增殖的活性较弱。

Gustin 等[38]以SKI-II 为先导物,运用拼合策略,通过将酚羟基替换为不同结构的环状氨基醇片段、改变氨基醇片段N 原子与苯环之间的距离、将氯原子替换为其他取代基等多种修饰策略,考察取代基电性、体积、位置等因素对活性的影响,通过多轮优化得到的化合物47 和48 均对SphK1有较强的抑制活性。
分析化合物47 与SphK1 的结合模式可以看出,该化合物与鞘氨醇采取相同的构象结合在J型口袋中,六元环状氨基醇末端可以与SphK1 相应的氨基酸残基产生氢键作用。其中,哌啶环的4-位羟基与Asp81 的羧基距离2.8 Å,与Asp81 残基的羰基发生氢键作用,2-位的羟甲基与Asp178 的羧基距离2.5 Å,与鞘氨醇的3-位羟基相似,与Asp178 发生氢键作用。同时,2-位羟甲基还可以通过水分子介导与Ala339、Gly342、Asp341、Ser168发生氢键作用,而Asp81、Asp178、Ser168 等都是影响SphK1识别底物的关键氨基酸残基(图2)。
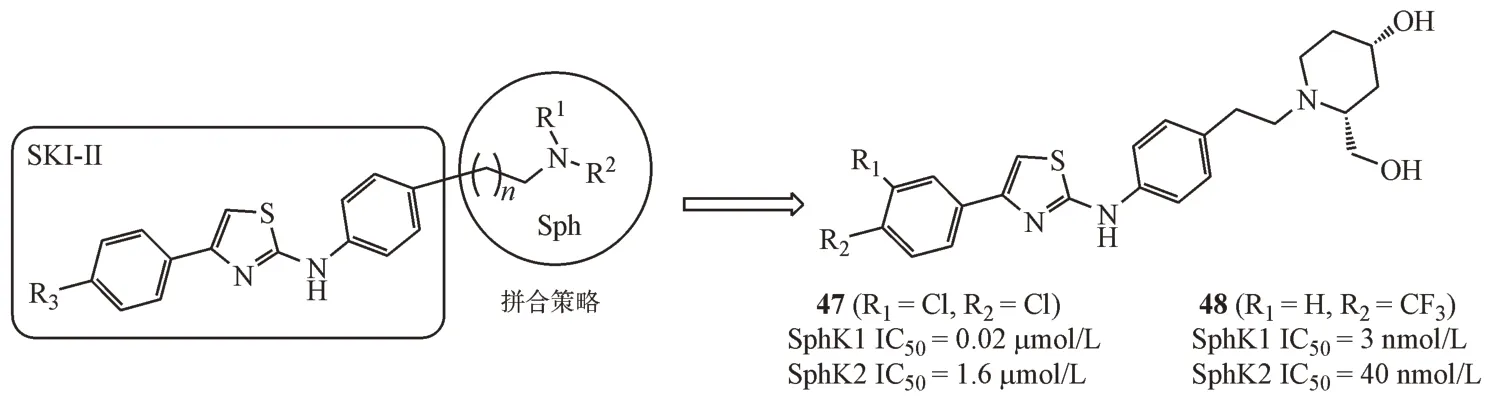

图2 化合物47与SphK1的结合模式
2.5 金刚烷类
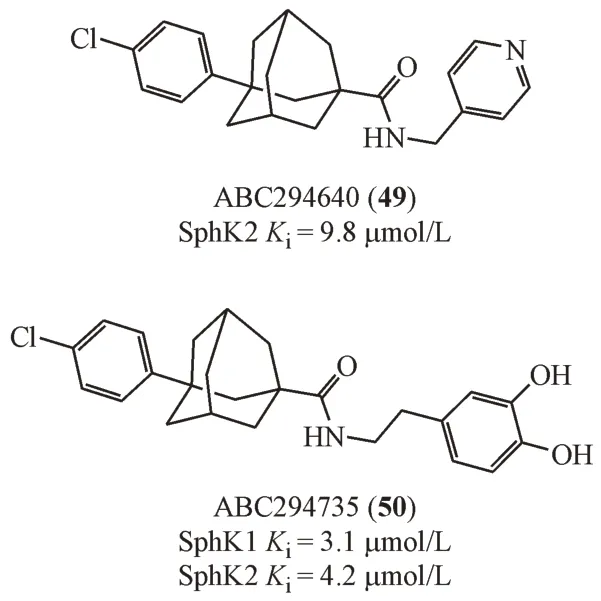
French 等[39]从化合物库中筛选到一个具有金刚烷骨架结构的鞘氨醇激酶抑制剂,进而合成了一系列金刚烷类衍生物,其中ABC294640(49)因具有较高的口服生物利用度而受到关注。ABC294640 作为选择性SphK2 抑制剂(Ki= 9.8 μmol/L),对包括肺癌、结肠癌、卵巢癌等多种实体肿瘤都有抑制作用。此外,ABC294640 对胰腺癌细胞也有很强的诱导凋亡作用,并且能够增强其对化疗的敏感性。目前ABC294640 在欧洲以孤儿药形式被批准治疗特定类型胰腺癌,治疗白血病的研究已进入临床Ⅱ期阶段。将ABC294640 的吡啶基团替换为邻苯二酚基团得到化合物ABC294735(50)是一个SphK1 和SphK2 的双重抑制剂。虽然这两个化合物抑制SphK 亚型的选择性有所区别,但是体内都观察到具有抗肾癌和胰腺癌增殖的作用,提示在该结构基础上开发SphK1抑制剂的可能性[40]。
2.6 ATP竞争性抑制剂
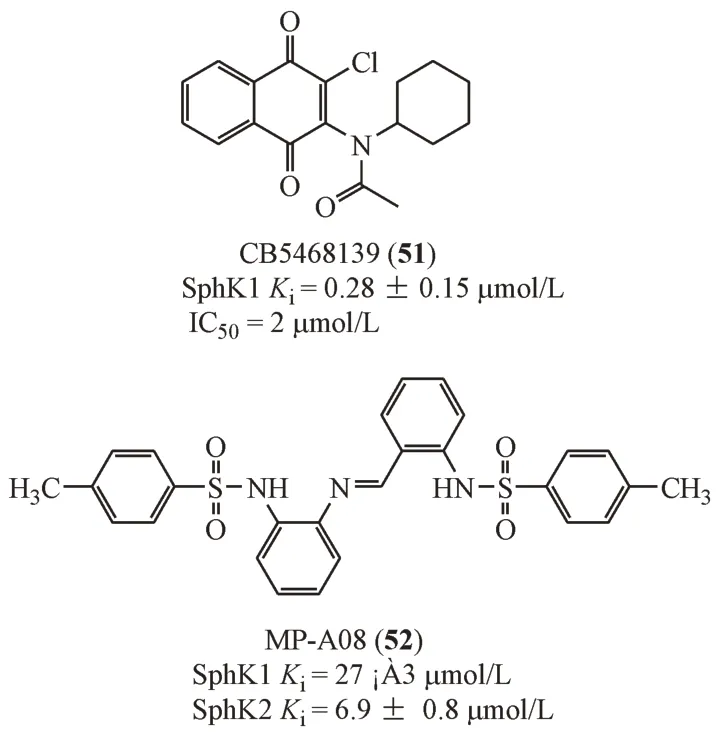
化合物51是首个被报道的以ATP结合位点为靶点的SphK1 抑制剂,虽然化合物51 对于SphK2没有抑制活性,但是,广泛的激酶测试数据表明,2 μmol/L浓度下该化合物对65种激酶中的12种抑制率在50%以上,显示出多激酶抑制效果[41]。
Pitman 等[42]用2 个细菌类脂质激酶(DgkB 和Yegs)进行同源模建,模拟SphK1 的ATP 结合位点结构,对超过10 万个化合物进行虚拟筛选得到MP-A08(52),其在细胞水平能够促进肿瘤细胞(A549、BJ7、MCF-7、MDA-MB-231)的Cer-Sph-S1P平衡向Cer方向偏移,诱导肿瘤细胞凋亡。在移植人肺癌细胞A549 的小鼠模型上观测到化合物52能有效地抑制肿瘤细胞的增殖并阻断肿瘤组织血管的生成。该化合物并不具有抑制其他激酶的活性,提示了在该结构基础上进一步研究选择性SphK1抑制剂的良好前景。
2.7 天然产物来源的抑制剂
Kono等[43]从天然产物中分离得到了一批结构新颖的SphK1 抑制剂。其中,化合物53 是从真菌Discomycete中分离得到的SphK 底物竞争抑制剂,其抑制SphK1 和SphK2 的IC50分别为41 和11 μmol/L。Bonhoure 等[44]发现化合物53 对于耐药性白血病时具有潜在的临床应用价值,对于多重耐药的急性髓细胞白血病,可以抑制肿瘤细胞HL-60内S1P 的生成,提高Cer 的含量,诱导肿瘤细胞凋亡。在化合物53 的作用下,慢性粒细胞白血病肿瘤细胞对伊马替尼的耐药性显著下降。化合物54是Kono 等[45]从海洋细菌SANK 71896中分离得到的SphK 底物竞争抑制剂,其抑制SphK1 和SphK2的IC50分别为7.8 和5.0 μmol/L,研究发现化合物54 可以增强前列腺癌细胞LNCaP 和PC-3 对多西他赛和喜树碱的敏感性。此外,Kono 等[46]还从真菌Zopfiella inermis中分离得到化合物55 和56,抑制小鼠肝组织SphK 的IC50分别为2.5 和1.6 μmol/L。

Jaspine B(57)是从海绵动物体内分离得到的天然产物,从结构上可以看做是鞘氨醇分子内脱水的类似物,能够抑制多种肿瘤细胞的增殖。构效关系研究显示,结构中四氢呋喃环的3个手性中心,其构型对鞘氨醇激酶抑制活性有比较大的影响,以化合物58 和59 的相对构型活性较好,脂肪链的长度以14个碳原子的长度为宜(60,61)[47]。

3 总结与展望
鞘脂及其代谢产物是生物体内重要的活性分子,参与细胞的生长、分化和衰老相关的信号转导过程。鞘氨醇激酶在鞘脂变阻器的动态平衡发挥重要作用,目前对于SphK1 亚型的生物学和抑制剂研究较为深入。已报道的SphK1 抑制剂包括鞘氨醇底物的结构修饰物、环状氨基醇类、脒基或胍基极性末端的化合物,也包括通过高通量筛选、天然产物分离提取得到的新结构分子。总体而言,SphK1 抑制剂的结构多样性尚未被充分挖掘。而且,通过检索德温特专利数据库发现,SphK1 和SphK2 抑制剂相关的公开不到300 项,表明该类抑制剂的结构研究和专利保护仍有较大空间。SphK1抑制剂可以促进肿瘤细胞凋亡,提高化疗药敏感性、降低或延缓耐药性,是抗肿瘤药物的优良靶标,而SphK2 抑制剂作为抗肿瘤药物、抗炎药物开发的关注度也逐渐提高。因此,进一步提高SphK1/ SphK2 选择性或者开发SphK 抑制剂并提高对其他激酶的选择性都是可以尝试的思路。目前SphK1的晶体结构已经被解析,抑制剂与SphK1的作用模式研究比较深入,SphK2的结构和与抑制剂的作用模式还需要进一步研究。因此,可以从SphK1 的三维结构出发,针对其三维结构观察到的,底物鞘脂与ATP 位点在空间位置上有所重叠的特点,开展基于结构的药物设计,尝试获得底物及ATP 均有结合作用的抑制剂,从而提高SphK1的选择性,或者提高SphK 与其他激酶的选择性,阐明抑制剂结构与活性、选择性的变化规律,研究其在体内的作用机制,充分挖掘该类抑制剂针对重大疾病的治疗价值。
