Next Steps for Efficacy Evaluation in Clinical Trials of COVID-19 Vaccines
2021-12-25HuDhunJingLiZhngJingXinLiFengCiZhu
Hu-Dhun Jing, Li Zhng, Jing-Xin Li, Feng-Ci Zhu,b,
a School of Public Health, Southeast University, Nanjing 210009, China
b NHC Key Laboratory of Enteric Pathogenic Microbiology, Jiangsu Provincial Center for Disease Control and Prevention, Nanjing 210009, China
c Center for Global Health, Nanjing Medical University, Nanjing 211166, China
d Department of Vaccine Clinical Evaluation, Jiangsu Provincial Center for Disease Control and Prevention, Nanjing 210009, China
1. Introduction
A vaccine clinical trial examines the effects of a vaccine on human volunteers in terms of safety,immunogenicity,and clinical efficacy through three distinct stages[1].In general,phase 1 studies focus on safety and reactogenicity, while phase 2 studies attempt to establish an immunogenicity proof of dose range,dosage, and immunization procedure (sometimes even efficacy data). Large phase 3 studies are designed to evaluate whether the dosing and vaccination schedule can deliver the desired protection efficacy with an acceptable safety profile [2]. A phase 3 vaccine clinical trial provides indispensable efficacy data to support a vaccine that has been issued with licensure.
To date,April 30,2021,27 coronavirus disease 2019(COVID-19)vaccines are being evaluated in phase 3 clinical trials, all of which are designed as individually randomized,placebo-controlled studies with 30 000–40 000 participants in each trial. The preliminary efficacies from the phase 3 trials of the ‘‘first wave” of ten COVID-19 vaccines were recently announced (Table 1): two mRNA vaccines (BNT162b2 developed by BioNTech/Pfizer, with a vaccine efficacy(VE)of 95.0%;and mRNA-1273 developed by Moderna,with a VE of 94.1%); four non-replicating viral-vectored vaccines(AZD1222 developed by the University of Oxford/AstraZeneca,with a VE of 70.0%; Sputnik V developed by Gamaleya, with a VE of 91.4%; Ad26.COV2.S developed by Janssen, with a VE of 66.9%;and Ad5-nCoV developed by CanSino Biologics and the Beijing Institute of Biotechnology, with a VE of 68.8%); a protein subunit vaccine (NVX-CoV237 developed by Novavax, with a VE of 89.3%); and three inactivated vaccines (BBIBP-CorV developed by the Beijing Institute of Biological Products, with a VE of 78.1%;the inactivated whole-virus nCov-19 vaccine developed by the Wuhan Institute of Biological Products, with a VE of 72.8%; and CoronaVac developed by Sinovac, with a VE of 91.3% in Turkey and 50.4% in Brazil) [3–8]. Based on these results, nine of the ten COVID-19 vaccines have been granted emergency-use authorization or conditional licensure in some regions or countries.Another protein subunit vaccine (ZF2001) has also received approval for emergency use.
As the COVID-19 pandemic continues to rage around the world,the demand for effective vaccines is an unprecedented huge. It is clear that the production amount of the first ten vaccines is unlikely to meet the world’s needs. Thus, research is still needed on other COVID-19 vaccine candidates, and will hopefully determine more effective vaccines against COVID-19 in phase 3 clinical trials.However, conducting individually randomized placebo-controlled clinical trials during a viral pandemic with such a high burden of disease and implementing an immunization campaign while previous approved vaccines are available have never been done before.The approval of the ‘‘first wave” of COVID-19 vaccines raises concerns about the administration of a placebo during the ongoing and future phase 3 trials of other COVID-19 vaccine candidates.
Here, we discuss the issues that may affect clinical trials of COVID-19 vaccines aiming to evaluate VE in the near future and examine the possibility of alternative trial designs, while taking ethical concerns,the circumstances of the epidemic,and statistical considerations into account.
2. Challenges for future COVID-19 vaccine clinical studies
2.1. Hundreds of COVID-19 vaccines under evaluation
As of April 30, 2021, 277 candidate vaccines against severe acute respiratory syndrome coronavirus 2(SARS-CoV-2)are under development worldwide, according to a survey by the World Health Organization (WHO) [9]. Among them, at least 93 vaccines have been approved for clinical trials. These approved vaccines mainly comprise protein subunit vaccines (n = 29, 31% of the 93 approved vaccines),viral vector vaccines(n=18,19%),inactivated vaccines(n=13,14%),DNA or RNA vaccines(n=23,25%),and viral particle vaccines (n = 5, 5%).
Aside from the ‘‘first wave” of COVID-19 vaccines, many of the vaccine candidates under development have as-yet undetermined efficacies;this is particularly true for the vaccines based on protein subunit technology platforms, which accounts for more than a third of the total. Such recombinant protein-based subunit vaccines are generally safe to administer to a wider variety of people,including children, seniors, and immune-compromised individuals;they are also easy to deal with in terms of storage and logistics.If all these vaccine candidates enter clinical trials at the same time,there may be insufficient resources to complete effective clinicaltrials, as was the case during the epidemic in the mainland of China, in the early part of the global pandemic, when trials for the development of COVID-19 therapeutic drugs lacked the ability to obtain solid evidence.
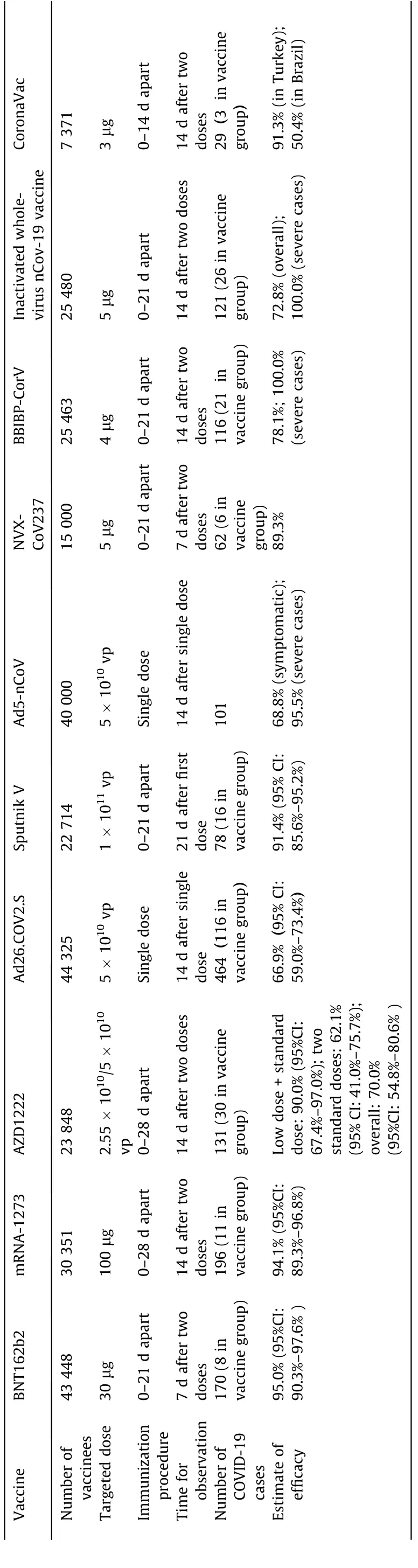
Table 1 Efficacy results of published COVID-19 vaccine candidates.
An efficacy trial should normally be carried out in a high-risk population that cannot access effective approved vaccines at that time, in areas experiencing an epidemic. However, conducting a clinical trial during an emergency situation posing huge challenges[10], especially if a vaccination campaign for emergency use has already been implemented that will reduce the pool of eligible study populations for subsequent COVID-19 vaccine trials. Therefore, the implementation of future phase 3 efficacy trials for COVID-19 vaccines may be concentrated in developing countries or regions with inadequate healthcare resources, which could impose a further burden on local healthcare systems and exacerbate the uneven accessibility of efficacious vaccines.
2.2. Limitations of ongoing COVID-19 vaccine clinical studies
The interim analysis results of the efficacy data from the phase 3 clinical trials of the ‘‘first wave” of COVID-19 vaccines have demonstrated their safety and efficacy against COVID-19,but over a fairly short term. The average observation periods for COVID-19 cases surveillance in the trials of BNT162b2, mRNA-1273, and AZD1222 were 2–3 months [3–5], so the durability of the protection and the long-term safety of these vaccines still remain to be determined. Due to the limited observation period, only a few or several severe COVID-19 cases were captured during the trials, leading to a lack of robust efficacy data on severe cases for these vaccines. In addition, although a similar VE was observed across subgroups defined by age in the open results of the phase 3 efficacy trials of BNT162b2, mRNA-1273, Sputnik V and Ad26.COV2.S [3,4,8,11], this data is not solid, as the small number of seniors enrolled in the trial and the protective efficacy of other candidate vaccines in seniors were not estimated. Therefore, the evidence on the protective effect of these vaccines in seniors is still insufficient. However, due to the good immunogenicity of the candidate vaccines in seniors, and to seniors’ high risk of being attacked by SARS-CoV-2, people aged 60 years and over are also eligible for vaccine administration in China.
As a result of all these knowledge gaps,scientists have called for the continuation of clinical trials with placebos,in order to avoid a loss of valuable research data and a decrease in health equity if participants do not belong to the identified priority groups [12].
3. Study design for VE evaluation
3.1. When placebo use is acceptable
According to the recommendations of a WHO expert panel,placebo use in vaccine trials is clearly acceptable when no efficacious and safe vaccine exists and when the vaccine under consideration is intended to benefit the population among which the vaccine is to be tested [13]. Although some COVID-19 vaccines have been approved for emergency use or conditional marketing, they are not considered to have the same level of known efficacy and safety as non-emergency vaccines for other diseases,since their approval was only based on short-term efficacy and safety data. Therefore,their emergency-use approval is not a marketing license,and many experts still advocate a placebo-use control in vaccine trials(Table 2).
3.1.1. Cross-over design
In a traditional (2 × 2) cross-over design, each participant receives two different treatments, which are labeled as A and B.Half of the participants receive A first;then,after a suitably chosen period of time, they cross over to receive B. The remaining participants receive B first, and then cross over to receive A. The aim of such a study is to compare the effects of A and B [14,15].This cross-over design could be an alternative method to evaluate the efficacy of COVID-19 vaccines in phase 3 trials, instead of placebo-controlled randomized trials (Fig. 1). During the first period of time, the participants would be individually randomized into a vaccine or placebo arm of the study; they would respectively receive shots of the vaccine or placebo, and then be followed up with to surveil for COVID-19 cases. After that, the participants in the vaccine group would cross over to receive the placebo, while the participants in the placebo group would receive the vaccine.Both the investigators and the participants would be masked to the group allocation. This design can maintain the benefits of placebo control and randomization without demanding an exceptional degree of altruism from the participants; in this way, it can help preserve the public’s trust that the scientists and regulators are prioritizing both the science and the participants [16].However, this design cannot be used to evaluate the long-term safety and efficacy of a vaccine compared with a placebo. Another concern is that offering the vaccine to the participants in the placebo arm would decrease health equity, because the participants in the placebo arm might not be prioritized for vaccination outside of the trial under conditions of vaccine scarcity.
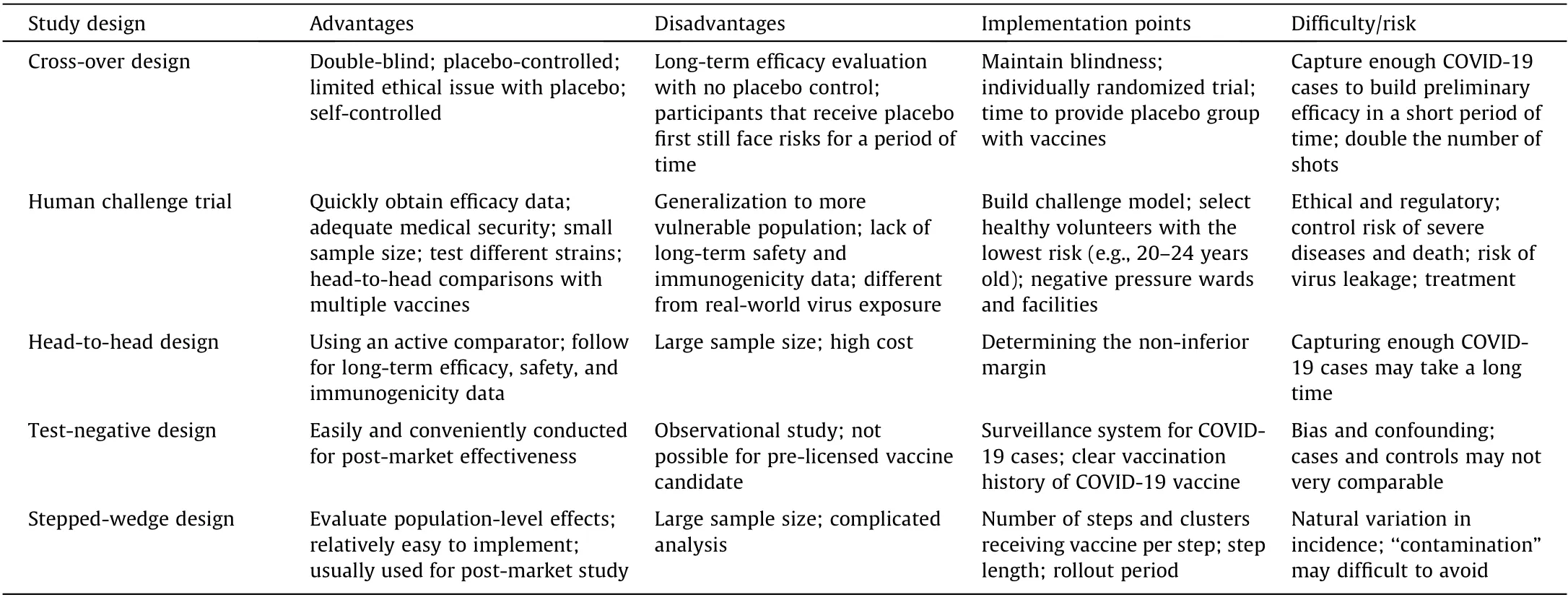
Table 2 Elements of the proposed study designs.
3.1.2. Human challenge trial
Human challenge trials involve the intentional infection of healthy, adult, consenting volunteers with an infectious agent(e.g.,a virus,parasite,bacteria,or fungus).The participants are randomly allocated to the vaccine or placebo arm of the study. After receiving intervention, the participants are intentionally exposed to the infectious agent in a controlled setting with adequate medication. The challenge strain should be well characterized,frequently attenuated, and manufactured under current good manufacturing practice (cGMP) or GMP-like conditions [17,18].Human challenge trials can move faster than traditional human trials and can quickly obtain VE data;furthermore,they need a much smaller sample size than a traditional randomized controlled trial.However, this design cannot evaluate VE against serious disease and death, because the challenge dose of the virus is carefully designed and the volunteers are treated early if they get sick.Nevertheless, in a human challenge trial for a COVID-19 vaccine, the volunteers in the placebo group may still face risks because there are no reliable treatments for severe COVID-19 [19]. Furthermore,the results obtained from young volunteers might not be reasonably extrapolated to seniors.In addition,the infectious agent used to challenge the volunteers may differ from a natural agent in terms of the dose or genes[19].
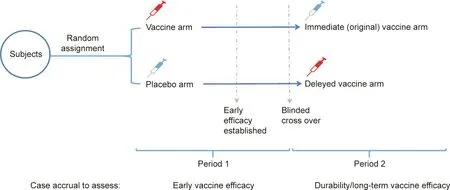
Fig. 1. Individual-randomized, double-blinded, and cross-over design for the efficacy evaluation of COVID-19 vaccines.
3.2. When placebo use is unacceptable
Placebo use in vaccine trials is clearly unacceptable when ①a highly efficacious and safe vaccine exists and is currently accessible in the public health system of the country in which the trial is planned,and ②the risks to the participants of delaying or foregoing the available vaccine cannot be adequately minimized or mitigated(e.g.,by providing counselling and education on behavioral diseaseprevention strategies, or by ensuring adequate treatment for the condition under study to prevent serious harm)[13].The data that accumulates as time goes by would support the vaccines that have already been approved for emergency use and have been demonstrated to be safe and effective.Thus,once the authorized vaccines become available in sufficient quantities to begin immunizing broader groups, it may no longer be feasible or ethical to include individuals in placebo-controlled trials [12]. Therefore, it is necessary to discuss alternative study designs without a placebo arm.
3.2.1. Head-to-head non-inferiority trial
A head-to-head non-inferiority randomized controlled trial aims to demonstrate that a new vaccine is no worse than an active comparator that has already shown its efficacy over a placebo within a prespecified margin that might be adopted when placebo use is not acceptable[20].Non-inferiority designs are useful in situations in which the efficacy of a new vaccine is deemed to be the same as that of the active comparator, but the new vaccine has additional benefits,such as fewer adverse events or reduced costs.A new vaccine is called‘‘non-inferior”to the comparator if the difference between the active comparator and the new vaccine is greater than the predefined margin (i.e., the boundary of the confidence interval exceeds the margin [20,21]). If the non-inferiority hypothesis is well established,it is even possible to perform a further superiority test on the active comparator, particularly for an active vaccine with only a moderate efficacy against clinical endpoints. However, compared with a placebo-controlled trial, a non-inferiority trial requires a larger sample size and a longer time to obtain clinical endpoints, since both arms of the study receive the vaccines, and the incidence of cases would be significantly reduced. Although the calculated sample size for a non-inferiority trial may vary from 20 000 to 50 000 people per year according to the different efficacies of the active comparator and the margin, it is still feasible.Moreover,a non-inferiority trial could also facilitate direct safety comparisons between the new vaccine and its established comparator [22].
3.2.2. Test-negative design
The test-negative design, which is a modified case-control study, has been introduced to assess vaccine effectiveness, especially against influenza vaccines[23].Under a test-negative design for vaccine effectiveness, the study subjects are all persons who seek medical care for suspected symptomatic illness. To evaluate COVID-19 vaccines,all subjects would first be tested using a highly specific assay(e.g.,polymerase chain reaction(PCR))for the detection of COVID-19; they would then be grouped according to their test results as either cases (those that test positive) or controls(those that test negative). Vaccine effectiveness would be estimated from the ratio of the odds of vaccination among the cases to the odds of vaccination among the controls [24]. From a practical standpoint, the test-negative design is easier to conduct than other study designs and makes it possible to minimize confounding due to health-care-seeking behavior [25]. However,a test-negative design is an observational study, so its validity depends on a careful assessment of potential biases and adjustment for them—particularly in terms of differences in disease severity among cases and non-cases.
3.2.3. Stepped-wedge design
A stepped-wedge cluster randomized controlled trial is commonly used for the evaluation of service delivery or policy interventions delivered at the level of the clusters. Examples of clusters may include schools, communities, factories, or families,although there are many other possible choices.In order to capture the population-level effects of an intervention, such as a vaccine designed to reduce the transmission of an infectious agent, a cluster randomized design can be adopted [26]. The design includes an initial period in which no clusters are exposed to the candidate vaccine. Subsequently, at regular intervals (the‘‘steps”),one cluster(or a group of clusters)is randomized to cross from the control to the vaccination under evaluation.This process continues until all clusters are exposed to the vaccination. Data collection continues throughout the study, so that each cluster contributes observations under both pre- and post-vaccination periods [27].The intervention effect is determined by comparing the data points in the post-vaccination section of the wedge with those in the control section. The stepped-wedge design is considered to be advantageous compared with a traditional parallel design, as long as there is a prior belief that the vaccination will do more good than harm,and when the vaccination can only be implemented in stages due to logistical,practical,or financial constraints[28].The biggest potential problem in using a stepped-wedge design to evaluate COVID-19 VE is the variation in COVID-19 incidence over time and space. In addition, the sample size of the design may be 100 times greater than that of an individually randomized trial.Therefore,the stepped-wedge design is usually used to estimate vaccine effectiveness after a vaccine license has been obtained.
4. Perspective
Traditionallarge-scaleindividual-randomized,placebocontrolled trials are the basis of modern clinical decision-making and remain the most efficient way to obtain reliable results for novel vaccines [29], as long as the trial’s risk–benefit profile remains acceptable [30]. Since emergency-use authorization and conditional licensure are not full licensures, the WHO has suggested that it is ethically acceptable to continue the blinded follow-up of placebo recipients in existing studies and to continue to perform placebo-controlled trials in order to yield unbiased evidence for the next vaccine candidates[29].However,as an increasing amount of evidence on the safety and efficacy of the COVID-19 vaccines is obtained and as authorized vaccines become more widely available, the risk-benefit profile of a normal placebocontrolled trial will become unacceptable, and the compliance of the trial may be impacted by drop-outs or‘‘contamination.”Therefore, alternative strategies to evaluate those vaccines are needed.
A placebo-controlled cross-over design could be used as an alternative to replace common placebo-controlled trials by reducing the ethical concern regarding long-term risks in the participants receiving a placebo; however, the long-term efficacy of the vaccine compared with that of a placebo would not be obtainable.Human challenge trials can also be used to accelerate the placebocontrolled efficacy estimation of a vaccine, when the use of a placebo in a large population is no longer recommended. However,the human challenge model can only be performed in a small number of participants, which may not be adequate for vaccine authorization, as an extended safety cohort and an extended head-to-head immunogenicity cohort are demanded in order to ensure a vaccine’s safety, immunogenicity, and persistency.
Head-to-head non-inferiority randomized controlled trials are an alternative design for the evaluation of new candidate COVID-19 vaccines.A head-to-head comparative design targeting efficacy requires a relatively large sample size and longer surveillance period for COVID-19 cases, to ensure that sufficient evidence on the vaccine’s relative efficacy can be obtained in order for the vaccine to be authorized[31].In general,a VE evaluation based on clinical endpoints is costly and time consuming.This will be especially true for trials using an active vaccine as a comparator.
In contrast, an efficacy evaluation of a vaccine based on the immunological correlates of protection is much easier to operate;it also saves time by measuring the proportion of vaccinees who generate a particular immune response, without capturing the clinical events [32]. A serological study strategy using an immunological correlate combined with an extended safety cohort has been successfully applied to assess other vaccines, such as influenza vaccines. However, the immunological correlate of protection for COVID-19 might not be available within a short period of time, since no standard serum immunological tests have been established as yet [33].
In order to evaluate vaccine effectiveness in the real world,nonrandomized observational studies, such as a test-negative design,are suggested.However,observational studies could yield misleading answers about safety and effectiveness, mainly due to the different risk exposures between vaccinated people and unvaccinated people during an epidemic[34,35].A potential strategy for assessing the population-level vaccine effectiveness against COVID-19 is to design cluster randomized trials that could reveal indirect effectiveness [36]; for example, a stepped-wedge approach has been recommended for use in evaluating vaccines in outbreak settings[37].
Nevertheless, post-licensure studies on vaccine effectiveness and safety in a large population are extremely important, particularly in order to screen for all rare severe adverse reactions. Some adverse events that have been reported in phase 3 studies,such as acute hypersensitive reaction, Bell facial paralysis, transverse myelitis,and abnormal plasma glucose,but whose association with COVID-19 vaccines has not been confirmed, will continue to be monitored and evaluated in post-marketing studies [38].
In conclusion, there remains a need to further evaluate the COVID-19 vaccines that were first authorized for use in order to fill in the knowledge gaps, and to develop additional vaccines that may be preferable for reasons of safety, efficacy, subgroup advantages, or logistics. Placebo-controlled trials are critical for efficacy and safety evaluation, and therefore should not be immediately eliminated.However,alternative trial designs must also be considered when the use of a placebo becomes ethically unacceptable and difficult to implement. With mass vaccination coverage anticipated, the findings from post-marketing data on COVID-19 vaccines can guide regulatory decisions and public health practices to maintain a positive benefit-risk balance.
杂志排行

Engineering的其它文章
- Visible Light-Induced 3D Bioprinting Technologies and Corresponding Bioink Materials for Tissue Engineering: A Review
- Temporal Profiles of Antibody Responses, Cytokines, and Survival of COVID-19 Patients: A Retrospective Cohort
- The East–West Divide in Response to COVID-19
- COVID-19 Vaccine Allocation: Modeling Health Outcomes and Equity Implications of Alternative Strategies
- Facilities for Centralized Isolation and Quarantine for the Observation and Treatment of Patients with COVID-19
- Non-Communicable Diseases During the COVID-19 Pandemic and Beyond