Mitochondrial Genome of Episesarma lafondii (Brachyura: Sesarmidae) and Comparison with Other Sesarmid Crabs
2021-12-22ZHANGYingGAOYanGONGLiLUXintingJIANGLihuaLIUBingjianLIULiqinZhenmingandLIPengfei
ZHANG Ying, GAO Yan, GONG Li, , *, LU Xinting, JIANG Lihua,LIU Bingjian, LIU Liqin, LÜZhenming, and LI Pengfei
Mitochondrial Genome of(Brachyura: Sesarmidae) and Comparison with Other Sesarmid Crabs
ZHANG Ying1), 2), GAO Yan1), 2), GONG Li1), 2), 3), *, LU Xinting1), 2), JIANG Lihua1), 2),LIU Bingjian1), 2), LIU Liqin1), 2), LÜZhenming1), 2), and LI Pengfei3)
1),,,316022,2),,316022,3),,530007,
Complete mitochondrial genomes (mitogenomes) can provide useful information for phylogenetic relationships, gene rearrangement, and molecular evolution. Here, the complete mitogenome of(Brachyura: Grapsoidea: Sesarmidae)was sequenced through next-generation sequencing technique for the first time.The 15640bp mitogenome contains the entire set of 37 genes and an AT-rich region.The rearrangements of two tRNA genes (-and-) are compared with that in the pancrustacean ground pattern, and the tandem duplication/random loss model was selected to explain the observed gene rearrangements. The phylogenetic results showed that all sesarmid crabs belong to the same group, wherein the genusshow- ed the closest relationship with. Furthermore, the monophyly of each family was well supported except for Xanthidae, Gecarcinidae, and Homolidae. The correlation between the phylogeny of Sesarmidae species and the gaps in theregion was analyzed. Evidently, the gaps betweenand(Gap3) and betweenand(Gap4) degenerated with the evolution process. In general, the results will contribute to the in-depth understanding of gene rearrangements in Sesarmidae mitogenomes and provide new insights into the phylogeny of Brachyura.
mitochondrial DNA; sesarmid crab; gene order; rearrangement mechanism; phylogenetic construction
1 Introduction
The mitochondrial genome (mitogenome) of metazoans is generally a closed, circular molecule with a size of 14–20kb.This genome contains 37 encoding genes, including 13 protein-coding genes (PCGs), two ribosomal RNA genes (and), 22 transfer RNA genes (tRNAs), and an AT-rich region (also called control region, CR) (Boore, 1999). The mitogenome is characterized with small genome size, high evolutionary rate, simple structure, and maternal inheritance (Gyllensten., 1991; Sato and Sato, 2013).The complete mitogenome has been widely used in com- parative genomics, adaptive evolution, population genetics, and phylogenetic studies (Plazzi., 2016; Tan., 2018; Tan., 2019; Irisarri., 2020). The mitogenome can also provide direct molecular clues for gene re- arrangement process, which will reveal important informa-tion for phylogenetic analyses (Liu and Cui, 2010; Zhuang and Cheng, 2010; Xin., 2017b; Gong., 2020a). With the rapid advancement of sequencing technologies, next-generation sequencing has become a fast and low-costmethod to provide complete mitogenomes (Gan., 2014; Tan., 2015).
The gene order was initially considered conserved in vertebrate mitogenomes. However, with more than 11000 complete mitogenomes currently determined, mitogenomic gene rearrangements have been found in several groups, including fishes (Lü., 2019), reptiles (Liu., 2019),birds (Caparroz., 2018), amphibians (Jiang., 2020),insects (Li., 2019a), and crustaceans (Tan., 2019). In general, the frequency of gene rearrangement in vertebrate mitogenomes is relatively lower than that in invertebrate mitogenomes. Thus far, several models have been applied for mitochondrial gene rearrangement.These mo- dels include the tandem duplication/random loss (TDRL) model (Moritz and Brown, 1987), recombination model (Lunt and Hyman, 1997), tRNA mispriming model (Can- tatore., 1987; Jacobs., 1989), tandem duplication/ nonrandom loss model (Lavrov., 2002), and double- replication/random loss model (Shi., 2014). The TDRL model posits that rearrangements occur via tandem dupli- cation followed by the random deletion of redundant genes (Moritz and Brown, 1987).This model has been widely used to explain the translocation of genes encoded on the same strand (Shi., 2015; Tan., 2018; Wang., 2018). Another commonly accepted hypothesis is the recombination model, which is characterized by the breakage and rejoining of the participating DNA strands (Lunt and Hyman, 1997). This model has been applied to explain the gene inversion and other rearrangement events (Kong., 2009; Tan., 2018). Usually the last three mo- dels are seldom used.
The infraorder Brachyura contains about 7250 described species, and most of them are economically and ecologi- cally prominent (Zhang, 2011; Basso., 2017; Chen.,2018). However, the phylogenetic relationships among members of Brachyura and their evolutionary origin remain debatable due to the extreme morphological and ecologi- cal diversity(Sanchez., 2016; Rocha., 2018; Wang., 2019). Brachyura was initially segmented into three groups: Podotremata, Heterotremata, and Thoracotremata (Spears., 1992). Subsequently, it was clustered intoDromiacea and Eubrachyura,which also include Thoraco- tremata, Raninoida, and Heterotremata) (Martin and Davis,2001). However, the latest classification scheme groups Bra-chyura into Eubrachyura, Dromicea, Raninoida, and Cyclo- dorippoida (Ahyong., 2007; Tsang., 2014). Al- though the phylogenetic relationship within Brachyura is still inconclusive, the current classification system has beensupported by most scholars.
Among Brachyura, sesarmid crabs are thought to be the key initial processors of leaves of mangrove, which play a crucial role in mangrove ecosystems (Gillikin, 2004; Ra- hayu and Ng, 2010; Tan., 2019). According to WoRMS(http://www.marinespecies.org/), the family Sesarmidae has36 genera and 309 species in total. Recently, studies on the genusde Man, 1895 (Brachyura: Sesarmidae) mainly concentrated on the morphology (Buatip., 2017; Jeyachandran., 2020), whereas studies at the mole- cular level are rare (Fratini., 2005; Schubart., 2009). To date,ten complete mitogenomes of Sesarmidae involving five genera are available from the National Cen- ter for Biotechnology Information (NCBI); however, no stu-dy reported the genus.Hombron & Jacquinot, 1846 (Sesarmidae, Sesarminae), which is a mudflat crab that is often found in the upper in- tertidal zone of mangrove forests in the Ryukyu Archipe- lago (Miyake., 2019). To date, no information is avail-able about its biological and molecular characteristics. Thus, in this study, the complete mitogenome of the genus() was sequenced for the first time. Thecharacteristics of this mitogenome were described and com-pared with those of 10 other sesarmid crabs.Additionally, anoverall phylogenetic analysis of 107 brachyuran species wascarried out based on the nucleotide sequences of 13 PCGs.
2 Materials and Methods
2.1 Ethics Statement
The crab specimens used in the present study were pur- chased from the Lingshui seafood market in Hainan, Chi- na. The species is not included in the endangered list of theInternational Union for Conservation of Nature (https:// www.iucnredlist.org/). Specimen collection and maintenance were performed in strict accordance with the recommen- dations of Animal Care Quality Assurance in China. All ex-perimental protocols were approved by the Institutional Ethics Committee of Zhejiang Ocean University.
2.2 Sample Collection and DNA Extraction
An individual specimen ofwas collected from Lingshui seafood market in Hainan Province, China (18˚24΄39΄΄N, 109˚58΄20΄΄E). The specimen was immediately preserved in absolute ethanol after collection and then stored at −20℃. This specimen was identified with a stereo dissecting microscope based on the key morphological features of crabs (Dai and Yang, 1991; Lee., 2015). The SQ Tissue DNA Kit (OMEGA) was used to ex-tract the total genomic DNA following the manufacturer’s instructions.
2.3 Mitogenome Sequencing and Assembly
The genomic DNA was sent to Shanghai Origingene Biopharm Technology Co., Ltd. for library preparation and high-throughput sequencing. The library was constructed by using the VAHTS Universal Plus DNA Library Prep Kit with an insert size of 400bp. The genome sequencing was conducted on an Illumina NovaSeq 6000 platform, with a strategy of 150 paired-end sequencing.The raw data sets were stored in the Short Read Archive database (https:// www.ncbi.nlm.nih.gov/sra/) with the accession no. SRX7 809611.The NOVOPlasty software (Dierckxsens., 2017) was used for theassembly of clean data without sequencing adapters.In the seed extension algo- rithm achieved by NOVOPlasty, the complete mitogenome of(GenBank accession number: NC_047209) was used as the seed sequence. To assess the single-base accuracy of the assembled genome, we com- pared it with three confirmed sequences by polymerase chain reaction (PCR) and Sanger sequencing methods.Sanger sequencing was conducted using an ABI genetic analyzer (Applied Biosystems, China). The PCR fragmentsand the corresponding primer sequences are as follows:(MW428280; F: TCNACAAAYCATAAAGAYATY GG/R: TANACYTCWGGRTGHCCRAARAAYCA);(MW430771; F: TRTGTGGRTTYCCHTTTHTAGCNGG/ R: GCTAATGCAG GGATACTAAC);(MW430770; F: TGRTTYGGRGCYTGRVTHGGNYT/R: GGDGGHA RNCCHCCWARNGA).
2.4 Mitogenome Annotation and Sequence Analyses
The software Sequin (version 15.10, http://www.ncbi. nlm.nih.gov/Sequin/) was used to manually annotate the complete mitogenome.The PCGs were determined by their open reading frame following the invertebrate mtDNA translation table. The boundaries of rRNA and tRNA geneswere determined using NCBI-BLAST (http://blast.ncbi.nlm.nih.gov) and tRNAscan-SE 1.21 (Lowe and Chan, 2016),respectively, comparing with the related species.Based on the secondary structure predicted by tRNAscan-SE 1.21 (Lowe and Chan, 2016) and MITOS Web Server (Bernt., 2013), we manually plotted the transfer RNA genes.The CGView online server V 1.0(Stothard and Wishart, 2005) was used to draw the mitogenome map. The soft- ware MEGA X (Kumar., 2018) was used to analyze the relative synonymous codon usage (RSCU) and nucleo- tide composition. The following formulas were used to cal- culate the strand asymmetries: AT-skew=(A−T)/(A+T); GC-skew=(G−C)/(G+C) (Perna and Kocher, 1995).
2.5 Phylogenetic Analysis
A total of 106 complete mitogenome sequences were downloaded from the GenBank database to reconstruct phy- logenetic relationships within Brachyura, adding two ano- muran species to serve as the outgroup (Table 1).Phylo- Suite (Zhang., 2019) was used to extract the nucleo- tide sequences of 13 PCGs for each of the above species from the GenBank files.The MAFFT program (Katoh., 2002) integrated into PhyloSuite was executed to align mul-tiple sequences in normal-alignment mode, and ambigu- ously aligned regions were identified and moved by Gblocks (Talavera and Castresana, 2007). The alignments of indi- vidual genes were then concatenated and used to generate input files (Phylip and Nexus format) for phylogenetic ana- lyses. GTR+F+I+G4 was selected as the best-fit model in accordance with the BIC criterion using ModelFinder (Kalyaanamoorthy., 2017). Phylogenetic trees were built under maximum likelihood (ML) and Bayesian in- ference (BI) methods. The ML analysis was carried out in IQ-TREE (Nguyen., 2015) using an ML+rapid boot- strap (BS) algorithm with 1000 replicates.The BI analysiswas performed in MrBayes 3.2.6 (Ronquist., 2012) with default parameters and 3×106Markov Chain Monte Carlo generations.The trees were sampled every 1000 ge- nerations with a burn-in of 25%. The average standard de- viation of split frequencies below 0.01 was considered to reach convergence.
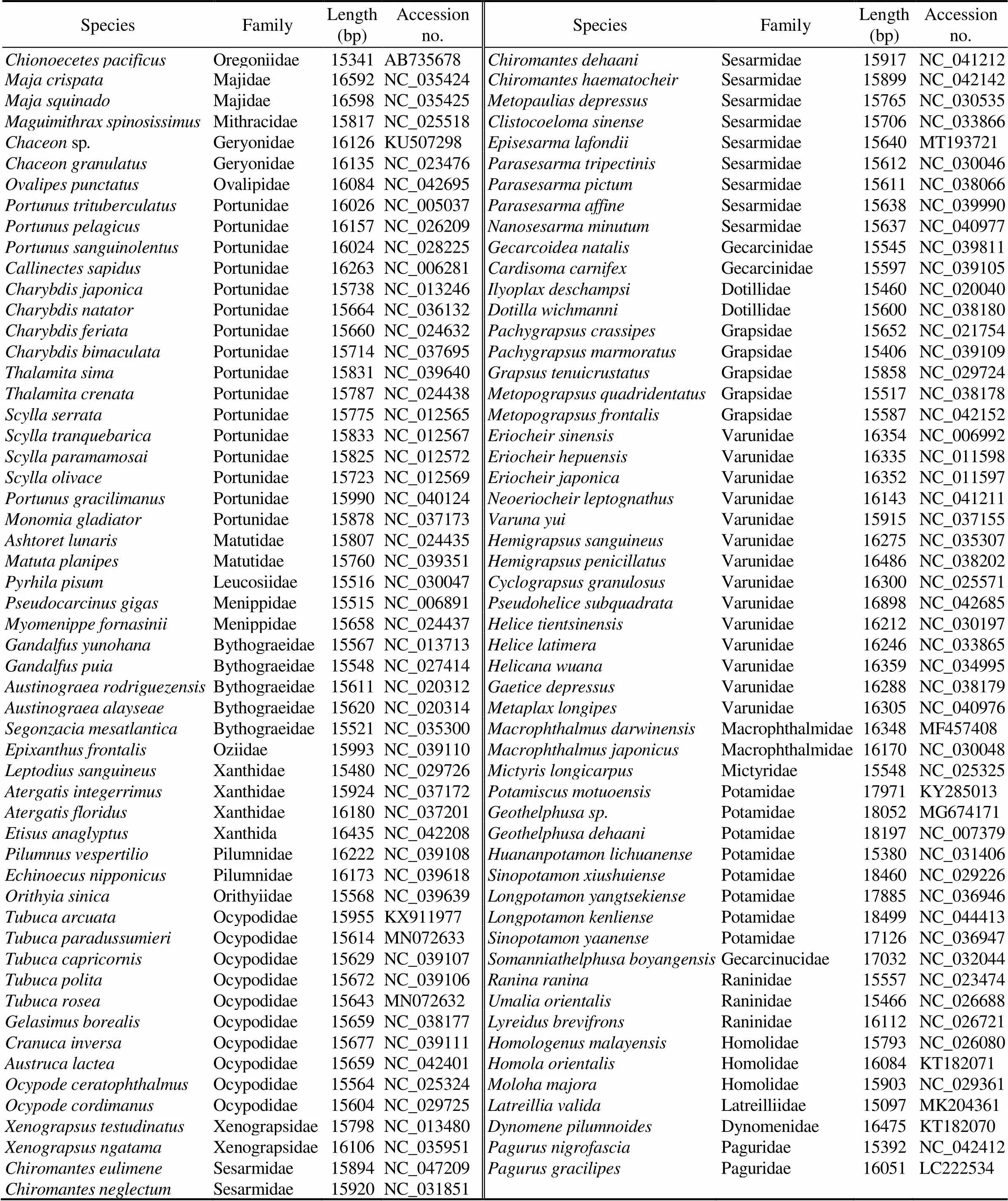
Table 1 List of 107 Brachyuran species and two outgroups used in this paper
3 Results and Discussion
3.1 Genome Structure and Composition
The circular duplex molecule mitogenome ofis 15640bp in size (GenBank accession number: MT193721), which is within the length range (15611–15920bp) of other previously sequenced Sesarmidae mitogenomes (Table 1). The mitogenome also contains 13 PCGs (11187bp), two rRNAs (2160bp), 22 tRNAs (1479bp), and a putative CR (663bp) (Fig.1; Table 2). The lengths of PCGs, tRNAs, rRNAs, and CR for this speciesand other published Sesar-midae species were compared. The sizes of PCGs, tRNAs, and rRNAs were relatively conserved, and the maximum length diversification, which is from 528bp to 833bp, was detected in the rapidly evolving CR.
The nucleotide composition of the whole mitogenome was 37.0% A, 38.9% T, 9.4% G, and 14.7% C (Table 3), which revealed a strong AT bias (75.9%). The skewness me-trics of the mitogenome showed negative AT-skew (−0.025)and negative GC-skew (−0.219), which followed the trendof published Sesarmidae mitogenomes (Table 4) (Xin.,2017a; Wang., 2018; Chen., 2019; Wang., 2019).Inmitogenome, 17 intergenic spacers ranging from 1bp to 38bp were found. The longest one was located betweenand(Table 2).Mean- while, 20bp overlapping sites were identified at eight junc- tions. Three overlaps (4bp betweenand, 1bp betweenand, and 7bp betweenand) were also generally found in other invertebrate species (Ta- ble 2) (Li., 2019b; Wang., 2020b).
3.2 PCGs and Codon Usage
The 13 PCGs consist of one cytochrome b (), two ATPases (and), three cytochrome c oxidases (-), and seven NADH dehydrogenases (-and). Four genes (,,, and) are encoded by the L-strand, whereas the remaining nine genes are encoded by the H-strand. The typical ATN co- dons are used as a start codon. The majority of the 13 PCGs terminate with TAA or TAG, whereas three other PCGs (,, and) use a single T as the stop codon (Ta- ble 2). The presence of different stop codons has been pro-ven to be a common phenomenon in metazoan mitoge- nomes (Wu., 2014; Hamasaki., 2017; Gong.,2018). The AT-skew of the 13 PCGs was negative (−0.161), whereas the GC-skew was positive (0.018), demonstrat- ing that Ts were more plentiful than As and Gs were more plentiful than Cs in the entire PCG sequence. Furthermore, four PCGs (,,, and) had positive values, indicating that they are encoded by the L-strand, whereas the remaining nine PCGs had negative values, in- dicating that they are encoded by the H-strand (Table 3).

Fig.1 Gene map of the E. lafondiimitogenome. Genes outside the circular are encoded by the heavy strand; genes inside the circular are encoded by the light strand.

Table 2 Features of the mitochondrial genome of E. lafondii
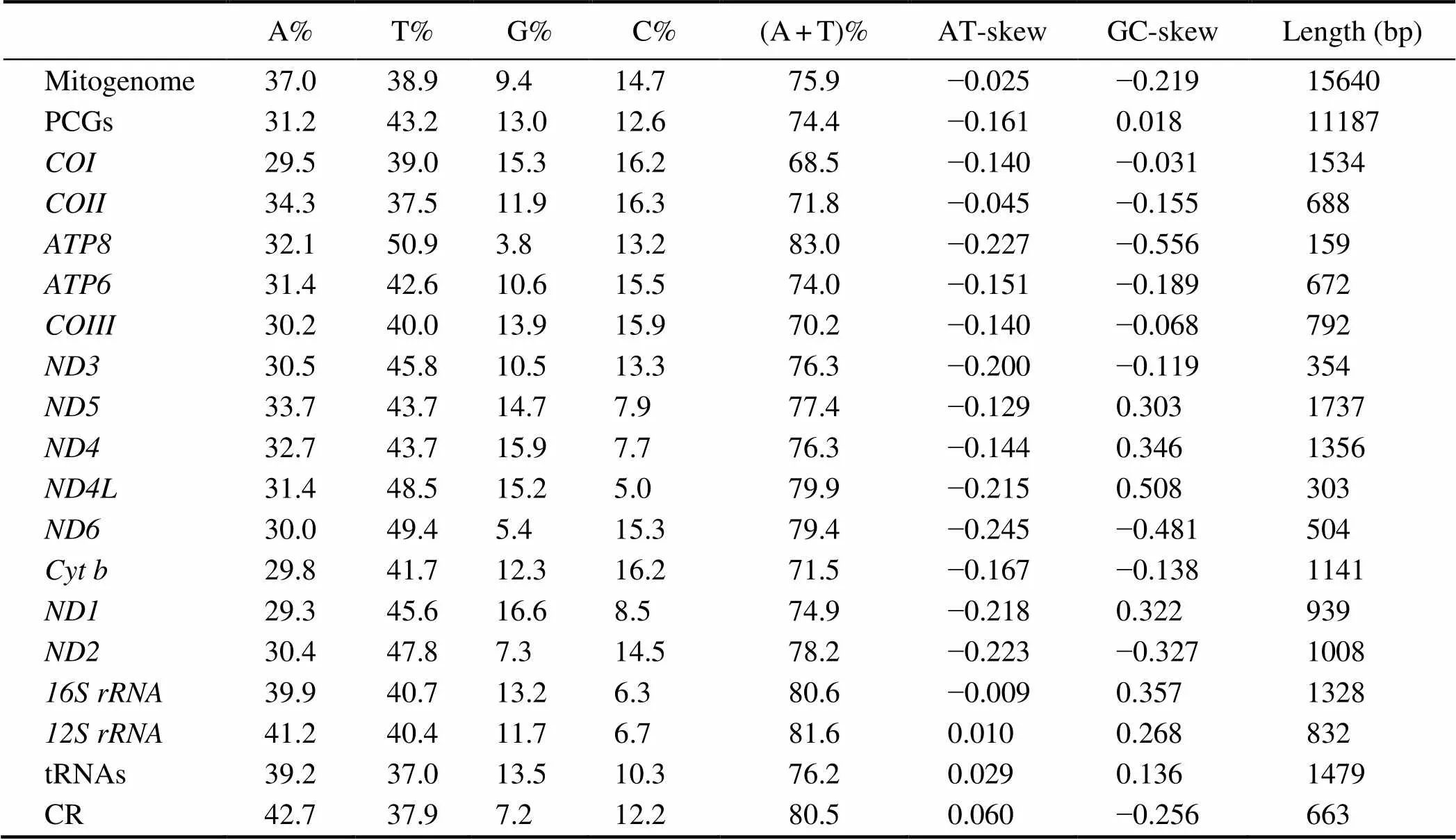
Table 3 Composition and skewness of E. lafondii mitogenome
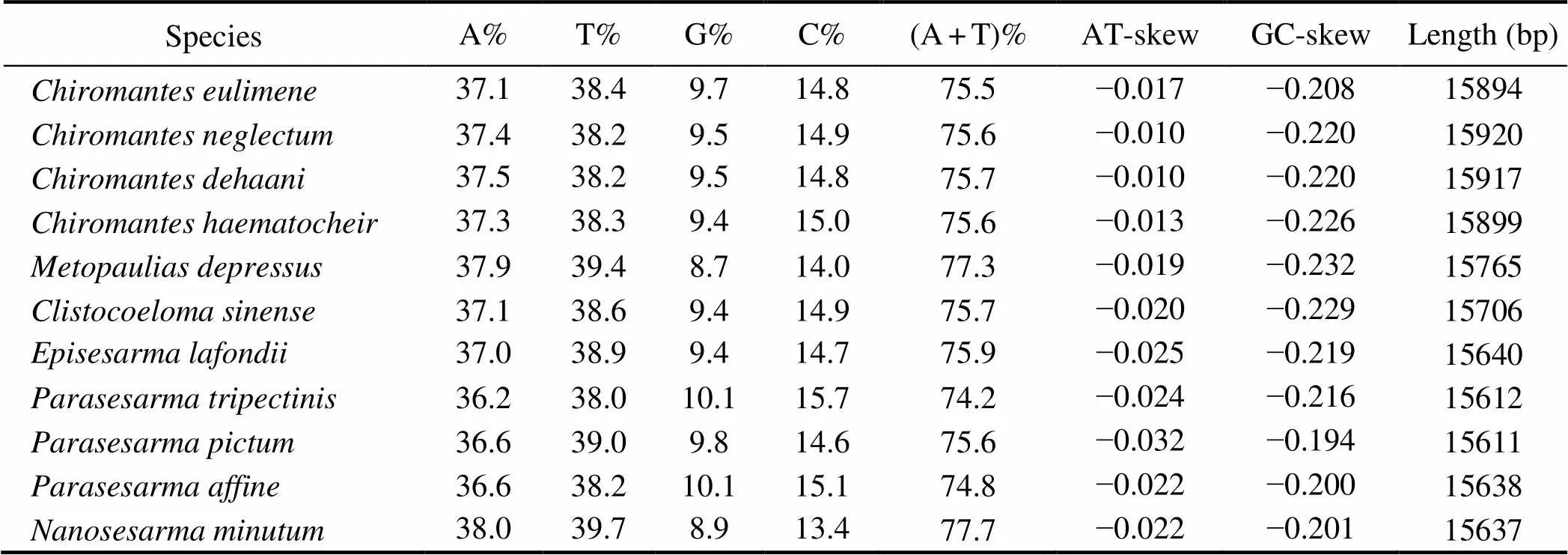
Table 4 Composition and skewness of mitogenomes in 11 Sesarmidae species
The RSCU and amino acid composition were highly si- milaramong the 11 Sesarmidae mitogenomes. Similar to other reported brachyuran species (Tang., 2018; Lu., 2020; Wang., 2020b), the usage of two- and four- fold degenerate codons in all sesarmid crabs was biased to-ward the use of codons abundant in A or T. The most com-mon amino acids were,,, andin all 11 Sesar- midae mitogenomes. In addition, the codons(CGC) and(UGC) are absent inandmitogenomes, whereas(CGC) is absent in the other five Sesarmidae species,including,,,,and.
3.3 Transfer RNAs, Ribosomal RNAs, and CR
A total of 22 tRNAs, ranging in length from 64bp to 73bp, were identified inmitogenome.Fourteen tRNAs are encoded by the H-strand, and the other eight tRNAs are encoded by the L-strand (Fig.1 and Table 2).Except for-(TCT), all tRNAs displayed canoni- cal cloverleaf structures. The loss of the dihydrouridine arm in(TCT) was thought to be a common pheno-menon in metazoan mitogenomes (Gong., 2019, 2020b).Except for the Watson-Crick base pairs (A-T and G-C) andG-U matches, four mismatched base pairs, including oneC-U base pair in, one A-A base pair in,and two U-U base pairs inand(L), were found. Posttranscriptional RNA editing may be involved in the correction of mismatches (Lavrov., 2000; Masta and Boore, 2004). Theandgenes are encoded by the L-strand, which are 1328 and 832bp, respectively. The location ofwas between(L) and, andwas located betweenand CR (Fig.1 and Table 2). The AT content of total rRNA was 81.0% (Table 3), indicating a highly AT preference.
The CR was located betweenand, with an extremely high AT content (80.5%). This region is 663bp in length and exhibits a positive AT-skew (0.060) and a negative GC-skew (−0.256) (Table 3). The CR was the most variable region because of its rapid evolution rate compared with the other genes. Thus far, limited research has studied the conserved blocks of the CR despite its func- tional importance, especially in invertebrate mitogenomes (Ray and Densmore, 2002; Guo., 2003; Zhao., 2011). Totally 11 Sesarmidae CRs were aligned to explore the sequence conservation. The results revealed nine con- served blocks (Fig.2), and their consensus sequences are as follows:AATGTA,ATATT, TTA,TAT,TTACTAT,ACCTGA ATT,TT, TTAATATATT(the underlined letters represent the fluctuant nucleo- tide among the 11 Sesarmidae species). To our knowledge, this is the first study to report the conserved blocks of the CR in crab mitogenomes.
3.4 Gene Rearrangement
Compared with the gene arrangement in ancestral crus- taceans (the pancrustacean ground pattern), the() was rearranged from the downstream of(Fig.3A) to the position between() and(), forming a new gene block (----) inmitogenome (Fig.3B).This translocation was also ob- served in other brachyuran mitogenomes (Chen., 2019; Tan., 2019; Wang., 2020b). However, an addi- tional translocation of() was identified when selecting the ancestral mitochondrial gene order of Bra- chyura as a reference. This translocation moved out from the() and() junction and formed a new gene cluster (---), in accord with otherpublished Sesarmidae mitogenome orders (Fig.3C) (Tan., 2019).
In accordance with the rearrangement features and prin- ciple of parsimony, the TDRL model was selected as the most suitable model to explain the two rearrangement events in themitogenome. The hypothesized interme- diate steps were as follows, starting with the typical an- cestral order of the Decapoda mitogenome. First, the gene block (--) was tandemly duplicated and generated two sets of the same gene cluster (--)- (--).Given the parsimony of the mitogenome, one of the duplicated genes lost function followed by a random loss of redundant genes, namely,--H-F′-ND5′-′ (the un- derlined letters represent the deleted genes, similarly he- reinafter). Thus, newgene order was formed (Fig.3B). In the second rearrangement event, the gene or- der of the gene cluster (--) was changed to--through the same mechanism. Thus, a dimeric (--)- (--) was formed due to gene duplication. In the fol- lowing step, the duplicated genes were deleted due to func- tional incapacitation (-----). Thus, a new gene order--was formed (Fig.3C).

Fig.2 Aligned sequences of the CRs in 11 sesarmid crabs. The shaded blocks represent the conserved sequences. Abbreviations of species names are given as follows. C. eul, Chiromentes eulimene; C. neg, Chiromantes neglectum; C. deh, Chiromantes dehaani; C. hae, Chiromantes haematocheir; M. dep, Metopaulias depressus; C. sin, Clistocoeloma sinense; E. laf, Episesarma lafondii; P. tri, Parasesarma tripectinis; P. pic, Parasesarma pictum; P. aff, Parasesarma affine; N. min, Nanosesarma minutum.

Fig.3 Inferred intermediate steps for the generation of the mitogenome of E. lafondii. A, Ancestral gene arrangement of Decapoda; B, Ancestral gene arrangement of Brachyura; C, Gene arrangement of E. lafondii and ten other Sesarmidae species. The duplicated gene block is underlined, and the lost genes are marked in gray.
In the above-speculated process, after the two copied gene clusters (--and--) lost their functions (losses 1, 2, 3, 4, and 5 in Fig.3), they would have de- graded to form five pseudogene fragments or short inter- genic spacers (gray boxes in Fig.3). Here, four intergenic spacers (Gaps 1, 2, 3, and 4) were found in themitogenome (the intergenic spacer between CR andde- faults to zero because the boundary of CR is uncertain): loss 1 () corresponds to Gap1 (5bp); loss 2 () to Gap2 (23bp); loss 4 () to Gap3 (38bp); loss 5 () to Gap4 (13bp). In general, given the high degradation rate of non-functional genes, the intergenic spacers caused by a random loss event should vanish rapidly to guarantee the parsimony of the mitogenome. The one-to-one correspon- dence between the loss-of-function fragments and residualintergenic spacers indicate that the novel gene order can be explained by the TDRL model.
3.5 Phylogenetic Analysis and QIM Spacers in Sesarmidae
The concatenated set of the nucleotide sequences of 13 PCGs from 107 known brachyuran species and two anomu- ran outgroups (and) were used for the phylogenetic analysis. The phylogenetic trees ob- tained using BI and ML methods resulted in identical topo- logical structures except for supporting values. Here, only one topology (BI) with both support values was presented (Fig.4). The phylogenetic tree showed that all Sesarmidaespecies clustered together as a group, wherein the genusshowed the closest relationship with. Sesarmidae and Gecarcinidae were the most closely related species, forming part of the superfamily Grapsoi- dea.Previous findings and our recent research on Sesarmi- dae species showed that the evolution process of mitoge- nomes could be revealed by the length of gap spacer in therearranged area (McKnight and Shaffer, 1997; Gong., 2020a; Zhang., 2020). Consequently, we analyzed thecorrelation between the phylogeny of Sesarmidae species and the gaps in theregion. The results reconfirmed that the phylogenetic position of each sesarmid crab was significantly correlated with the gaps in the rearrangedregion(Fig.5).In this study, the gap spacer betweenand(Gap3) decreased from 218bp () (Zhang., 2020) to 14bp (). Thespacer betweenand(Gap4) followed the same trend, decreasing from 64bp () to 9bp ().This trend suggests that with the evo- lution of sesarmid crabs, the gap spacers (Gap3 and Gap4) decreased progressively.
Of the 29 families in our phylogenetic tree, except for Xanthidae, Gecarcinidae, and Homolidae, each family form- ed a monophyletic clade with high nodal support values (Fig.4).Thus it needs further attention with the morpho- logical identification and taxonomic status of their closely related species,.,,,, and. With the exception of Eriphioidea, Ocypodoidea, and Grapsoidea, the monophyly of most superfamilies were well supported, as consistently revealed in previous molecular phyloge- netic analyses (Tan., 2018; Tan., 2019; Lu., 2020; Wang., 2020a). For a long time, the classifica- tion of Grapsoidea and Ocypodoidea has been controver- sial.Previous studies based on morphological features con- sidered them to be monophyletic clades(Martin and Da- vis, 2001; Ng, 2008; Davie., 2015). However, an in- creasing number of molecular studies, including ours, havechallenged the monophyly of these taxa (Chen., 2018; Tan., 2018; Chen., 2019; Lu., 2020).Wang.’s recent molecular study revealed that Ocypodoidea and Grapsoidea are divided into three clades (Wang., 2020a), and similar findings are presented in Tan.’s work (2018, 2019). Although the polyphyly of Grapsoi- dea and Ocypodoidea is well supported,the phylogenetic relationships of these superfamilies need further analysis by integration of additional molecular data.
Although the main phylogenetic structures of our tree followed those of previous results, several controversial findings were observed.Here,Varunidae and Macrophthal- midae possessing the same gene order were clustered to- gether as sister groups and were distantly related to Mic- tyridae, which is consistent with most molecular viewpo- ints (Gong., 2018; Chen., 2019; Tan., 2019; Wang., 2020a). Meanwhile, in a recent study, the phy- logenetic relationship among the above three families was ((Macrophthalmidae+Mictyridae)+Varunidae) (Tan., 2018). Additionally, no agreement was obtained on the phy- logenetic relationships among Sesarmidae, Gecarcinidae, Dotillidae, and Grapsidae.Here, Sesarmidae and Gecarci- nidae were closely related, and Dotillidae was the sister clade to Grapsidae, supporting Chen.’s viewpoint (Chen., 2019). In the previous findings (Basso., 2017; Tan., 2018), Sesarmidae was clustered with Dotilli- dae as sister groups. Regarding the phylogenetic position ofGecarcinidae and Grapsidae, no consensus has been reach- ed in current studies (Sanchez., 2016; Jia., 2018; Tan., 2019). In general, the placement of multiple sin- gle brachyuran lineages in the tree may produce conflict- ing phylogenetic relationships, possibly affecting the position of other brachyuran clades at a high taxonomic le- vel.In our phylogenetic tree, most of the unstable and con- flicting clades might have resulted from the limited taxon samples.Thereby, more comprehensive taxon samplings andreliable classification markers are necessary for fur- ther understanding the phylogenetic and evolutionary rela- tionships among Brachyura.

Fig.4 Phylogenetic tree of brachyuran species inferred from the nucleotide sequences of 13 PCGs based on ML and BI analyses. The node marked with a solid circle indicates 100 ML bootstrap support (BS) and 100% BI posterior probability (PP). The numbers after the species name are the GenBank accession number.
4 Conclusions
In this article, the complete mitogenome ofis determined and analyzed for the first time. The molecular features of this newly sequenced mitogenome aremostly consistent with those of 10 other sesarmid crabs. The gene rearrangement events occurring inmitogenome can be explained by the TDRL model. Phylogenetic ana- lyses indicate the close relationship ofandand the non-monophyly of Xanthidae, Gecar- cinidae, and Homolidae.Moreover, the polyphyly of three superfamilies (Ocypodoidea, Eriphioidea, and Grapsoidea)is reconfirmed.In the future studies, more samples fromdifferent taxonomic levels and reliable classification mar- kers will be employed to facilitate the taxonomical and phy- logenetic studies of Brachyura.

Fig.5 Relationship of Sesarmidae species and gap spacers between tRNA-QIM. Gap3 and Gap4 indicate the intergenic spacer between Q and I, and between I and M, respectively.
Acknowledgments
This work was supported by the Natural Science Foun- dation of Zhejiang Province (No. LY21C190007) and the Zhoushan Science and Technology Bureau (No. 2021C21 007). We would like to express our gratitude to Dr. Xu Zhang for helping in species identification and providing critical comments. The valuable remarks of the anonymous reviewers are also acknowledged.
Ahyong, S. T., Lai, J. C. Y., Sharkey, D., Colgan, D. J., and Ng, P. K. L., 2007. Phylogenetics of the brachyuran crabs (Crustacea: Decapoda): The status of Podotremata based on small subunit nuclear ribosomal RNA., 45 (2): 576-586.
Basso, A., Babbucci, M., Pauletto, M., Riginella, E., Patarnello, T., and Negrisolo, E., 2017. The highly rearranged mitochon- drial genomes of the crabsand(Majidae) and gene order evolution in Brachyura., 7 (1): 1-17.
Bernt, M., Donath, A., Jühling, F., Externbrink, F., Florentz, C., Fritzsch, G.,., 2013. MITOS: Improvedmetazoanmitochondrial genome annotation., 69 (2): 313-319.
Boore, J. L., 1999. Animal mitochondrial genomes., 27 (8): 1767-1780.
Buatip, S., Thongroy, P., and Yeesin, P., 2017. Burrow morpholo- gical characteristics of(H. Milne Edwards, 1853) (Decapoda, Grapsidae, Sesarminae)., 22 (2): 17-30.
Cantatore, P., Gadaleta, M., Roberti, M. N., Saccone, C., and Wil- son, A. C., 1987. Duplication and remoulding of tRNA genes during the evolutionary rearrangement of mitochondrial genomes., 329 (6142): 853-855.
Caparroz, R., Rocha, A. V., Cabanne, G. S., Tubaro, P., Aleixo, A.,Lemmon, E. M.,., 2018. Mitogenomes of two neotropical bird species and the multiple independent origin of mitochon- drial gene orders in Passeriformes.,45 (3): 279-285.
Chen, J., Xing, Y., Yao, W., Xu, X., Zhang, C., Zhang, Z.,., 2019. Phylomitogenomics reconfirm the phylogenetic positionof the genusinferred from the two grapsid crabs (De-capoda: Brachyura: Grapsoidea)., 14 (1): e0210763.
Chen, J., Xing, Y., Yao, W., Zhang, C., Zhang, Z., Jiang, G.,.,2018. Characterization of four new mitogenomes from Ocypo-doidea & Grapsoidea, and phylomitogenomic insights into tho- racotreme evolution., 675: 27-35.
Dai, A. Y., and Yang, S. L., 1991.. Chi- na Ocean Press, Beijing, 482-495.
Davie, P. J., Guinot, D., and Ng, P. K., 2015.Systematics and clas-sification of Brachyura. In:. Brill, 1049-1130.
Dierckxsens, N., Mardulyn, P., and Smits, G., 2017. NOVOPlasty:assembly of organelle genomes from whole genome data., 45 (4): e18.
Fratini, S., Vannini, M., Cannicci, S., and Schubart, C. D., 2005. Tree-climbing mangrove crabs: A case of convergent evolution., 7 (2): 219-233.
Gan, H. M., Schultz, M. B., and Austin, C. M., 2014. Integrated shotgun sequencing and bioinformatics pipeline allows ultra- fast mitogenome recovery and confirms substantial gene re- arrangements in Australian freshwater crayfishes., 14 (1): 19.
Gillikin, D. P., 2004. Osmoregulatory ability of(Crosnier, 1965) subjected to dilute and hypersaline sea- water., 77 (1): 67-74.
Gong, L., Liu, B. J., Liu, L. Q., Guo, B. Y., and Lü, Z. M., 2019. The complete mitochondrial genome of(Cen- trarchiformes: Terapontidae) and comparative analysis of the control region among eight Centrarchiformes species., 45 (2): 137-144.
Gong, L., Lu, X., Luo, H., Zhang, Y., Shi, W., Liu, L.,., 2020a.Novel gene rearrangement pattern inmitochondrial genome: New gene order in genus(Pleuronectiformes: Cynoglossidae)., 149: 1232-1240.
Gong, L., Lu, X., Wang, Z., Zhu, K., Liu, L., Jiang, L.,.,2020b. Novel gene rearrangement in the mitochondrial genome of(Anomura: Coenobitidae) and phy- logenetic implications for Anomura., 112 (2): 1804- 1812.
Gong, L., Lü, Z. M., Guo, B. Y., Ye, Y. Y., and Liu, L. Q., 2018. Characterization of the complete mitochondrial genome of thetidewater goby,(Gobiiformes; Gobii-dae; Gobionellinae) and its phylogenetic implications., 10 (1): 93-97.
Guo, X., Liu, S., and Liu, Y., 2003. Comparative analysis of the mitochondrial DNA control region in cyprinids with different ploidy level., 224 (1-4): 25-38.
Gyllensten, U., Wharton, D., Josefsson, A., and Wilson, A. C., 1991. Paternal inheritance of mitochondrial DNA in mice., 352 (6332): 255-257.
Hamasaki, K., Iizuka, C., Sanda, T., Imai, H., and Kitada, S., 2017.Phylogeny and phylogeography of the land hermit crab(Decapoda: Anomura: Coenobitidae) in the Nor- thwestern Pacific Region., 38 (1): e12369.
Irisarri, I., Uribe, J. E., Eernisse, D. J., and Zardoya, R., 2020. A mitogenomic phylogeny of chitons (Mollusca: Polyplacopho- ra)., 20 (1): 1-15.
Jacobs, H. T., Herbert, E. R., and Rankine, J., 1989. Sea urchin egg mitochondrial DNA contains a short displacement loop (D- loop) in the replication origin region., 17 (22): 8949-8965.
Jeyachandran, S., Park, K., Kwak, I. S., and Baskaralingam, V., 2020. Morphological and functional characterization of circu- lating hemocytes using microscopy techniques., 83 (7): 736-743.
Jia, X. N., Xu, S. X., Bai, J., Wang, Y. F., Nie, Z. H., Zhu, C. C.,., 2018. The complete mitochondrial genome ofand phylogenetic analysis of Genus(Crustacea: Decapoda: Parathelphusidae)., 13 (2): e0192601.
Jiang, L., Zhang, M., Deng, L., Xu, Z., Shi, H., Jia, X.,., 2020.Characteristics of the mitochondrial genome ofand related species in Ranidae: Gene rearrangements and phylogenetic relationships., 10 (23): 12817-12837.
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A., and Jermiin, L. S., 2017. ModelFinder: Fast model selection for accurate phylogenetic estimates., 14 (6): 587-589.
Katoh, K., Misawa, K., Kuma, K. I., and Miyata, T., 2002. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform., 30 (14): 3059-3066.
Kong, X., Dong, X., Zhang, Y., Shi, W., Wang, Z., and Yu, Z., 2009. A novel rearrangement in the mitochondrial genome of tongue sole,: Control region transloca- tion and a tRNA gene inversion., 52 (12): 975-984.
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K., 2018. MEGA X: Molecular evolutionary genetics analysis across com- puting platforms., 35 (6): 1547-1549.
Lavrov, D. V., Boore, J. L., and Brown, W. M., 2002. Complete mtDNA sequences of two millipedes suggest a new model for mitochondrial gene rearrangements: Duplication and nonran- dom loss., 19 (2): 163-169.
Lavrov, D. V., Brown, W. M., and Boore, J. L., 2000. A novel typeof RNA editing occurs in the mitochondrial tRNAs of the cen- tipede.s, 97 (25): 13738-13742.
Lee, B. Y., Ng, N. K., and Ng, P. K., 2015. The taxonomy of five species ofDe Man, 1895, in Singapore (Crustacea: Decapoda: Brachyura: Sesarmidae)., 31 (Supplement): 199-215.
Li, N., Hu, G. L., and Hua, B. Z., 2019a. Complete mitochondrial genomes ofandand geno- mic comparisons of Mecoptera., 140: 672-681.
Li, W., Cheng, J., Hui, M., and Sha, Z., 2019b. Molecular phy- logeny of the genus(Decapoda: Anomura: Dio- genidae) based on mitochondrial and nuclear DNA sequences., 37 (5): 1686-1697.
Liu, J., Yu, J., Zhou, M., and Yang, J., 2019. Complete mitochon- drial genome of: Deep insights into the phy- logeny and gene rearrangements of Agamidae species., 125: 423-431.
Liu, Y., and Cui, Z., 2010. Complete mitochondrial genome of the Asian paddle crab(Crustacea: Decapo- da: Portunidae): Gene rearrangement of the marine brachyu- rans and phylogenetic considerations of the decapods., 37 (5): 2559-2569.
Lowe, T. M., and Chan, P. P., 2016. tRNAscan-SE On-line: Inte- grating search and context for analysis of transfer RNA genes., 44 (W1): W54-W57.
Lu, X., Gong, L., Zhang, Y., Chen, J., Liu, L., Jiang, L.,., 2020.The complete mitochondrial genome of: The first representative from the family Calappidae and its phylo- genetic position within Brachyura., 112 (3): 2516- 2523.
Lunt, D. H., and Hyman, B. C., 1997. Animal mitochondrial DNA recombination., 387 (6630): 247-247.
Lü, Z., Zhu, K., Jiang, H., Lu, X., Liu, B., Ye, Y.,., 2019. Complete mitochondrial genome ofreveals novel gene order and phylogenetic relationships of Anguilliformes., 135: 609-618.
Martin, J. W., and Davis, G. E., 2001.. Natural History Museum of Los An- geles County, Science Series, 39: 1-124.
Masta, S. E., and Boore, J. L., 2004. The complete mitochon- drial genome sequence of the spiderreveals rearranged and extremely truncated tRNAs., 21 (5): 893-902.
McKnight, M. L., and Shaffer, H. B., 1997. Large, rapidly evolv- ing intergenic spacers in the mitochondrial DNA of the sala- mander family Ambystomatidae (Amphibia: Caudata)., 14 (11): 1167-1176.
Miyake, T., Aihara, N., Maeda, K., Shinzato, C., Koyanagi, R., Kobayashi, H.,., 2019. Bloodmeal host identification with inferences to feeding habits of a fish-fed mosquito,., 9 (1): 1-8.
Moritz, C., and Brown, W. M., 1987. Tandem duplications in ani- mal mitochondrial DNAs: Variation in incidence and gene con- tent among lizards., 84 (20): 7183-7187.
Ng, P., 2008. Systema Brachyurorum, Part I. An annotated check- list of extant brachyuran crabs of the world., 17: 1-286.
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q., 2015. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies., 32 (1): 268-274.
Perna, N. T., and Kocher, T. D., 1995. Patterns of nucleotide com- position at fourfold degenerate sites of animal mitochondrial genomes., 41 (3): 353-358.
Plazzi, F., Puccio, G., and Passamonti, M., 2016. Comparative large-scale mitogenomics evidences clade-specific evolution- ary trends in mitochondrial DNAs of Bivalvia., 8 (8): 2544-2564.
Rahayu, D. L., and Ng, P. K. L., 2010. Revision of the(Latreille, 1803) species-group (Crustacea: Decapoda: Brachyura: Sesarmidae)., 2327 (1): 1-22.
Ray, D. A., and Densmore, L., 2002. The crocodilian mitochon- drial control region: General structure, conserved sequences, and evolutionary implications., 294 (4): 334-345.
Rocha, C. T., Regina, W. M., Mantelatto, F. L., Christopher, T., and José, Z. F., 2018. Ultrastructure of spermatozoa of mem- bers of Calappidae, Aethridae and Menippidae and discussion of their phylogenetic placement., 101 (1): 89- 100.
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Hohna, S.,., 2012. MrBayes 3.2: Efficient Ba-yesian phylogenetic inference and model choice across a large model space., 61 (3): 539-542.
Sanchez, G., Tomano, S., Yamashiro, C., Fujita, R., Wakabaya- shi, T., Sakai, M.,., 2016. Population genetics of the jum- bo squid(Cephalopoda: Ommastrephidae) in the northern Humboldt Current system based on mitochondrial and microsatellite DNA markers., 175: 1- 9.
Sato, M., and Sato, K., 2013. Maternal inheritance of mitochon- drial DNA by diverse mechanisms to eliminate paternal mito- chondrial DNA.–, 1833 (8): 1979-1984.
Schubart, C. D., Liu, H. C., and Ng, P. K. L., 2009. Revision ofSerène & Soh, 1970 (Crustacea: Brachyura: Sesarmi- dae), with description of a new genus and two new species., 2154 (1): 1-29.
Shi, W., Gong, L., Wang, S. Y., Miao, X. G., and Kong, X. Y., 2015.Tandem duplication and random loss for mitogenome rear- rangement in(Teleost: Pleuronectiformes)., 16 (1): 355.
Shi, W., Miao, X. G., and Kong, X. Y., 2014. A novel model of double replications and random loss accounts for rearrangements in the mitogenome of(Teleostei: Pleu- ronectiformes)., 15 (1): 352.
Spears, T., Abele, L. G., and Kim, W., 1992. The monophyly of Brachyuran crabs: A phylogenetic study based on 18s rRNÅ., 41 (4): 446-461.
Stothard, P., and Wishart, D. S., 2005. Circular genome visuali- zation and exploration using CGView., 21 (4): 537-539.
Talavera, G., and Castresana, J., 2007. Improvement of phyloge- nies after removing divergent and ambiguously aligned blocks from protein sequence alignments., 56 (4): 564-577.
Tan, M. H., Gan, H. M., Lee, Y. P., Bracken-Grissom, H., Chan, T. Y., Miller, A. D.,., 2019. Comparative mitogenomics of the Decapoda reveals evolutionary heterogeneity in architecture and composition., 9 (1): 1-16.
Tan, M. H., Gan, H. M., Lee, Y. P., Linton, S., Grandjean, F.,Bartholomei-Santos, M. L.,., 2018. ORDER within thechaos: Insights into phylogenetic relationships within the Ano- mura (Crustacea: Decapoda) from mitochondrial sequences and gene order rearrangements., 127: 320-331.
Tan, M. H., Gan, H. M., Schultz, M. B., and Austin, C. M., 2015. MitoPhAST, a new automated mitogenomic phylogeny tool in the post-genomic era with a case study of 89 decapod mito- genomes including eight new freshwater crayfish mitogeno- mes., 85: 180-188.
Tang, B. P., Liu, Y., Xin, Z. Z., Zhang, D. Z., Wang, Z. F., Zhu, X. Y.,., 2017. Characterisation of the complete mitochon- drial genome of(Grapsoidea: Varunidae) and comparison with other Brachyuran crabs., 110 (4): 221-230.
Tsang, L. M., Schubart, C. D., Ahyong, S. T., Lai, J. C. Y., Au, E. Y. C., Chan, T. Y.,., 2014. Evolutionary history of true crabs (Crustacea: Decapoda: Brachyura) and the origin of fresh-water crabs., 31 (5): 1173- 1187.
Wang, Q., Tang, D., Guo, H., Wang, J., Xu, X., and Wang, Z., 2020a. Comparative mitochondrial genomic analysis ofand insights into the phylogeny of the Ocypodoidea & Grapsoidea., 112 (1): 82-91.
Wang, Z., Shi, X., Guo, H., Tang, D., Bai, Y., and Wang, Z., 2020b.Characterization of the complete mitochondrial genome ofand comparison with other Brachyuran crabs., 112 (1): 10-19.
Wang, Z., Shi, X., Tao, Y., Wu, Q., Bai, Y., Guo, H.,., 2019. The complete mitochondrial genome of(Brachyura: Grapsoidea: Sesarmidae) and comparison with other Brachyuran crabs., 111 (4): 799-807.
Wang, Z., Wang, Z., Shi, X., Wu, Q., Tao, Y., Guo, H.,., 2018. Complete mitochondrial genome of(Brachyura: Sesarmidae): Gene rearrangements in Sesarmidaeand phylogenetic analysis of the Brachyura., 118: 31-40.
Wu, X., Xiao, S., Li, X., Li, L., Shi, W., and Yu, Z., 2014. Evo- lution of the tRNA gene family in mitochondrial genomes of fiveclams (Bivalvia, Veneridae)., 533 (1): 439- 446.
Xin, Z. Z., Liu, Y., Zhang, D. Z., Chai, X. Y., Wang, Z. F., Zhang, H. B.,., 2017a. Complete mitochondrial genome of(Brachyura: Grapsoidea): Gene rearrange- ments and higher-level phylogeny of the Brachyura., 7 (1): 1-10.
Xin, Z. Z., Yu, L., Zhang, D. Z., Wang, Z. F., Zhang, H. B., Tang, B. P.,., 2017b. Mitochondrial genome of(Brachyura: Grapsoidea: Varunidae): Gene rearrange- ments and higher-level phylogeny of the Brachyura., 627: 307-314.
Zhang, D., Gao, F., Jakovlic, I., Zou, H., Zhang, J., Li, W. X.,.,2019. PhyloSuite: An integrated and scalable desktop platformfor streamlined molecular sequence data management and evo-lutionary phylogenetics studies., 20 (1): 348-355.
Zhang, Y., Gong, L., Lu, X., Jiang, L., Liu, B., Liu, L.,., 2020.Gene rearrangements in the mitochondrial genome of(Brachyura: Sesarmidae) and phylogenetic im-plications for Brachyura., 162: 704-714.
Zhang, Z. Q., 2011..,3148 (Special issue): 1-237.
Zhao, L., Zheng, Z. M., Huang, Y., Zhou, Z., and Wang, L., 2011.Comparative analysis of the mitochondrial control region in Orthoptera., 50 (3): 385-393.
Zhuang, X., and Cheng, C. H. C., 2010. ND6 gene ‘lost’ and found:Evolution of mitochondrial gene rearrangement in Antarctic no- tothenioids., 27 (6): 1391- 1403.
September 25, 2020;
December 1, 2020;
April 27, 2021
© Ocean University of China, Science Press and Springer-Verlag GmbH Germany 2021
.E-mail: gongli1027@163.com
(Edited by Qiu Yantao)
杂志排行

Journal of Ocean University of China的其它文章
- Meshless Method with Domain Decomposition for Submerged Porous Breakwaters in Waves
- Facial Features of an Air Gun Array Wavelet in the Time-Frequency Domain Based on Marine Vertical Cables
- Magma Evolution Processes in the Southern Okinawa Trough:Insights from Melt Inclusions
- Summery Intra-Tidal Variations of Suspended Sediment Transportation–Topographical Response and Dynamical Mechanism in the Aoshan Bay and Surrounding Area, Shandong Peninsula
- High-Resolution Geochemical Records in the Inner Shelf Mud Wedge of the East China Sea and Their Indication to the Holocene Monsoon Climatic Changes and Events
- Geological Guided Tomography Inversion Based on Fault Constraint and Its Application