丙硫菌唑合成工艺改进
2021-12-18刘志勇姚文强池伟林
刘志勇 姚文强 池伟林
(1.江苏剑牌农化股份有限公司,江苏 盐城 224700;2.东阳光农药研究所,广东 东莞 523000)
丙硫菌唑(prothioconazole)是拜耳公司研制的一种广谱的三唑硫酮类杀菌剂,为甾醇脱甲基化(麦角甾醇生物合成)抑制剂,主要用于防治谷物、麦类豆类作物等众多病害。丙硫菌唑是一个商业化开发非常成功的产品,已成为全球杀菌剂中的领先产品。其2004年上市,2019年的全球销售额已达8.25亿美元。丙硫菌唑在国内的专利已经在2015年11月份过期,其硫化工艺专利也于2018年到期。其合成工艺是通过硫粉氧化饱和硫酮三唑环或者三唑芳香环得到,存在氧化不完全,废液含有硫污染大的问题。后续化学工作者们通过改进工艺,探索出了不少方法来解决这些问题,例如用双氧水或者氯化铁来代替硫粉。但是也存在着一些问题,比如反应中起氧化作用的双氧水的氧化能力太强而导致有大量脱硫杂质的产生,或者最后一步合成丙硫菌唑用的是氯化铁氧化,用量过大,存在废水污染大的问题。
随着国家对环境保护的重视程度和环境保护要求的日益提高,开发丙硫菌唑的清洁合成工艺具有非常重要的意义。我们通过不断改进,以alpa-乙酰基-gama-丁内酯为原料,经过磺酰氯氯化、盐酸开环、氢氧化钠关环、磺酰氯再氯化先得到中间体IV;然后用邻氯氯苄与镁反应得到的格氏试剂与IV反应得到中间体VI,中间体VI直接与水合肼发生肼解反应得到肼基化合物VII,最后直接关环并通空气直接氧化得到丙硫菌唑(见图1)。实现了从中间体VI合成VII的过程中,VI直接与水合肼反应,不需要先环氧化再与水合肼反应,并且降低了反应温度;从中间体VII合成硫菌唑VIII的过程中,直接用空气氧化,避免了用氯化铁、硫或者双氧水作为氧化剂,减少了废液的量,可以简化操作步骤,减少废液的产生,并且反应均为常规合成工艺,操作简单。

图1 丙硫菌唑的合成路线
1.实验部分
1.1 仪器与试剂
1H NMR在VARIAN Mercury-Plus 400核磁共振仪(Varian,Inc.,Palo Alto,U.S.A.)上测定,CDCl3为溶剂,并将TMS(四甲基硅烷)用作内标。多重性由以下缩写表示:s,单峰; d,双峰; t,三峰; m,多重峰; br,宽峰。液相色谱仪(Agilent 1260,美国安捷伦公司)。所有市售试剂均为分析纯,无须额外纯化。
1.2 实验部分
1.2.1 中间体I的合成

将alpa-乙酰基-gama-丁内酯(800 g, 6.24 mol)加入1000 mL三口瓶中,冷却至10℃。慢慢滴加磺酰氯(880 g, 6.52 mol),温度控制10℃,滴完搅拌一小时,用水泵抽干,加入水搅拌(300 mL*3),分液。得到1015g黄色油状物,产率定量。
1.2.2 中间体II的合成

在500 mL三口瓶中,加入I(1000 g, 6.15 mol),盐酸( 1250+1000 mL, 22 mol),氯化铝( 1.3 g, 0.01mol),加热回流反应2小时,进行水蒸气蒸馏,分液,有机相用食盐水洗(100 mL*3)。得到740 g黄色油状物,产率77.6%,纯度94.4%。
1.2.3 中间体III的合成

在500mL三口瓶中,加入水( 300 mL, 3.89 mol),氢氧化钠( 238.7 g, 5.97 mol),四丁基溴化铵(43.0 g, 0.133 mol),加热到100℃,慢慢滴加II(740 g, 4.77 mol),一小时滴完, 100℃反应2小时,进行水蒸气蒸馏,温度120-130℃,馏分分液,有机相用食盐水(100 mL*3)洗,干燥剂干燥,除去干燥剂得到420 g黄色油状物,产率74.3 %,纯度97.2%。
1.2.4 中间体IV的合成

将III(420 g, 3.54 mol),150mL甲醇加入1000 mL三口瓶中,冷却至0℃。慢慢滴加磺酰氯(660 g, 4.88 mol),温度控制低于10℃以下,三个小时滴完,滴完搅拌一小时,加入冰水搅拌洗涤三次(300 mL*3),饱和碳酸氢钠洗至中性,分液,有机相100-120℃减压精馏,合并纯度相似的馏分,得到448.89 g无色油状物2-氯-1-(1-氯环丙基)乙酮,产率82.8%,纯度93.17%。

1.2.5 中间体VI的合成

1L升三口瓶中,将镁粉(14.5g,0.6mol)和1.0 g碘分散于34ml甲苯和16ml甲基四氢呋喃混合溶剂,通氮气置换空气。缓慢滴加邻氯氯苄(81.5g,0.5mol)的甲苯(115ml)/甲基四氢呋喃(55ml)溶液,控制体系温度35-40℃。滴加完毕,室温搅拌3h, 取样中控V,格试剂含量>85%为反应合格。IV(85g,0.5mol)溶于甲苯(60ml)/甲基四氢呋喃(20ml),冰浴下缓慢滴入上述反应体系,控制体系温度小于15℃。滴完后升至室温搅拌2h。取样中控IV <0.5%为反应合格。冰浴下向上述体系滴加260ml10%的HCl,室温搅拌3h。加适量水洗涤,分液。有机相直接用于下一步反应。

1.2.6 中间体VII的合成
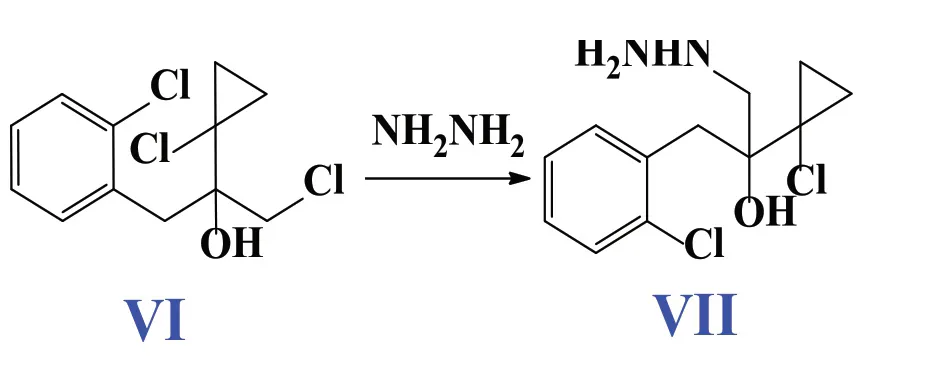
1L升三口瓶中, 加入上一步反应混合液,加入水合肼 (125g,98%,0.5mol),升温至70℃,搅拌12h,取样中控,VI<0.5%为反应合格。水洗,无水硫酸镁干燥有机相,过滤,直接用于下一步反应。


1.2.7 丙硫菌唑VIII的合成。
1L升三口瓶中, 加入上一步反应混合液,冰浴搅拌,加入60ml50%NaOH,升至室温,搅拌45min。加入甲醛 (21.2g,37%,262mmol)溶液,继续搅拌45min。
冰浴下向上述体系依次加入NaSCN(22.4g,278mmol),NH4SCN(7g,92.4mmol)和NaHSO4.H2O (85g,278mmol),搅拌的过程中不断通入空气,室温搅拌20h。分液,有机相以饱和氯化钠溶液洗涤,水洗,无水硫酸钠干燥。浓缩,正己烷沉淀,过滤,烘干。得浅灰色固体70g。产率69.5%。

2.结果与讨论
2.1 从中间体VI合成VII的过程中,温度对产率的影响
在生产中,一般要求温度不能太高,否则会浪费能源并且带来操作上的风险。在肼解反应中,由于中间体VI端基上的氯活性较低,因此一般需要超过90度的温度。我们通过不断筛选条件,直接将中间体VI与水合肼在无溶剂条件下反应,发现在70℃的时候,反应12小时能反应完全,即使提高温度,虽然能反应完全,却没必要。但是温度低于70℃,比如在60℃下反应的时候,还有一部分没反应完全,所以反应温度控制在70℃比较合适(见表1)。
中间体VII的结构可以通过核磁表征出来,从图2可以看出来,与苯基和肼基相连的亚甲基上的两个氢在其他基团的影响下裂分成了两个峰,分别在δ= 2.97, 2.67和δ=3.59, 3.42处,肼基上的氢则表现出一个宽的单峰,在δ=3.5-2.8之间,羟基也是一个大的宽峰,在δ=5.0-4.5之间。

图2 中间体VII的氢谱
2.2 合成丙硫菌唑的过程中,通空气时间对产率的影响
在传统工艺中,合成丙硫菌唑的最后一步是通常是用硫粉、氯化铁或者双氧水氧化,这些方法存在着污染大,或者副产物多的缺点。我们对现有工艺进行尝试,直接用中间体VII与甲醛和氰酸盐在乙酸乙酯中反应,并且不断通空气,发现反应所得的中间产物IX能直接转化为丙硫菌唑(见图3),并且能在20小时反应完全。

图3 合成丙硫菌唑的过程
从图4可以看出,在反应5小时的时候,已经生成了78.23%的丙硫菌唑,中间产物IX含量有16.84%;反应10小时的时候,丙硫菌唑的含量已经达到了89.26%,中间产物IX还剩下6.61%;反应20小时的时候,中间产物IX已经完成反应完成,丙硫菌唑的含量已经达到了95.18%。

图4 通空气时间对产率的影响
3.结论
在现有研究报道的基础上,结合实验过程,发现了VI合成VII的过程中,可以在较低温度下直接与水合肼反应,不需要先环氧化再与水合肼反应,节约了能源并提高了操作的安全性;VII合成丙硫菌唑的过程中,可以直接通空气氧化,在20小时内反应完全,避免了用氯化铁、硫或者双氧水作为氧化剂,减少了废液的量,使工艺更加清洁环保,且处理方便,收率高。
