4例B型儿童尼曼-匹克病的临床特点分析及文献复习
2021-12-08覃莹莹单庆文
覃莹莹 单庆文
【关键词】 尼曼匹克病;SMPD1基因;儿童
中图分类号:R596.1 文献标志码:B DOI:10.3969/j.issn.1003-1383.2021.10.014
尼曼-匹克病(Niemann-Pick disease,NPD)是一种罕见的常染色体隐性遗传病,属先天性糖脂代谢性疾病,其特点是脂类过量,主要是鞘磷脂和胆固醇累积于患者的肝脏、脾脏、肺脏、骨髓甚至脑部等重要器官,导致出现临床症状轻重不同的一组疾病。NPD可分为A、B、C三型,其中A型和B型均由编码酸性鞘磷脂酶(acid sphingomyelinase,ASM)的SMPD1基因突变导致,C型则因NPC1或NPC2基因突变导致。国外已有的研究数据显示,A/B型合并发病率在0.5/10万~1/10万[1]。为了提高对本病的认识,现总结广西医科大学第一附属医院收治的4名儿童尼曼-匹克病B型患者的临床资料,并结合文献复习,分析报道如下。
1 资料与方法
1.1 一般资料
收集广西医科大学第一附属医院2012年1月至2019年7月收治的4例NPD患儿资料,均为男性,就诊年龄1岁3个月~2岁7个月,病程为2个月~1年3个月,4例患者均为NPD-B型,均无家族史。
1.2 方法
收集4例患儿临床资料进行回顾性分析,包括年龄、性别、家族史、病程、临床表现、治疗方案、随访结果、血常规、肝功能、血脂分析、腹部B超、骨髓细胞形态学检查、基因检测等。
1.3 NPD-B型臨床诊断标准
对于肝脾肿大伴肝功能异常、血小板减少、间质性肺疾病、血脂异常,尤其是高密度脂蛋白胆固醇血清水平降低、低密度脂蛋白胆固醇升高及高甘油三酯血症的患者应高度怀疑NPD-B型。骨髓细胞学检查发现典型的尼曼匹克细胞可协助诊断,患者的酸性鞘磷脂酶活性(外周血淋巴细胞或皮肤成纤维细胞培养)低于正常值的10%或 SMPD1基因分析检出2个等位基因已知致病变异即可诊断为A/B型尼曼-匹克病,再根据是否出现神经系统症状来区分该病为NPD-A型还是NPD-B型。
2 结 果
2.1 临床表现
4例患者均有不同程度的肝脏、脾脏肿大,3例患者有腹部膨隆,3例患者表现为生长发育迟缓,3例患者表现有咳嗽,4例患者均无黄疸及肌张力异常。
2.2 实验室检查
4例患者均行血常规检查,白细胞降低1例,轻度贫血3例,血小板降低1例;4例患者均行肝功能检查,ALT升高3例,AST升高4例,总胆汁酸升高3例;4例患者行血脂检查,胆固醇升高2例,甘油三酯升高4例,低密度脂蛋白胆固醇升高2例,高密度脂蛋白胆固醇降低3例。
2.3 影像学检查
4例患者行腹部超声检查均提示肝脏、脾脏肿大,其中1例患者合并右肾萎缩。3例患者行胸部影像学检查(1例胸片,2例肺部CT),其中胸片表现为肺纹理粗,1例肺部CT表现为右肺上叶尖后段及两肺下叶背段炎症,1例肺部CT表现为两肺各叶见斑片状密度增高影及磨玻璃影,并可见小叶间隔增厚、边缘模糊,呈铺路石征。
2.4 骨髓细胞学检查
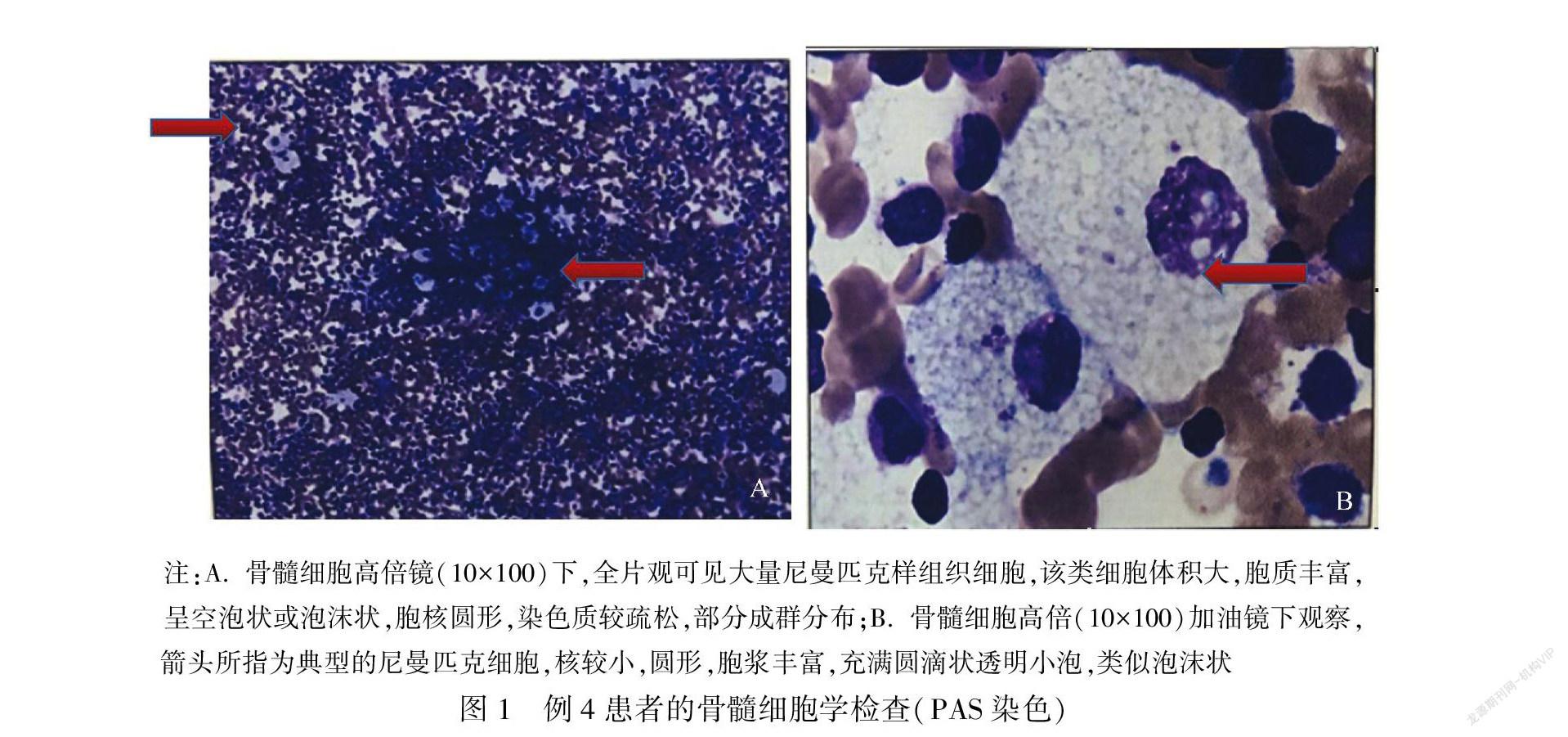
4例患者行骨髓细胞学检查,其中3例可找到尼曼匹克细胞(图1A、B),1例表现为反应性骨髓象(粒系成熟迟缓)。
2.5 肝脏组织病理检查
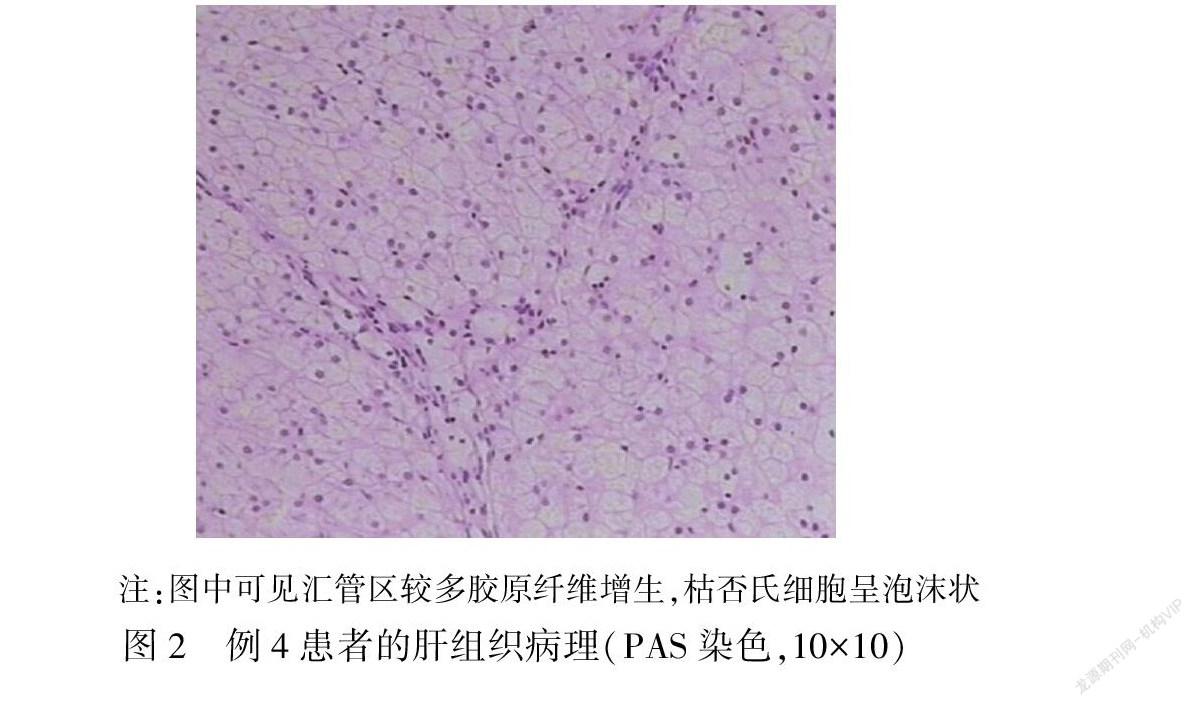
3例患者行肝脏组织穿刺活检,在病理检查中均发现泡沫细胞(见图2),肝脏组织病理均有不同程度的炎症和纤维化表现。
2.6 基因结果
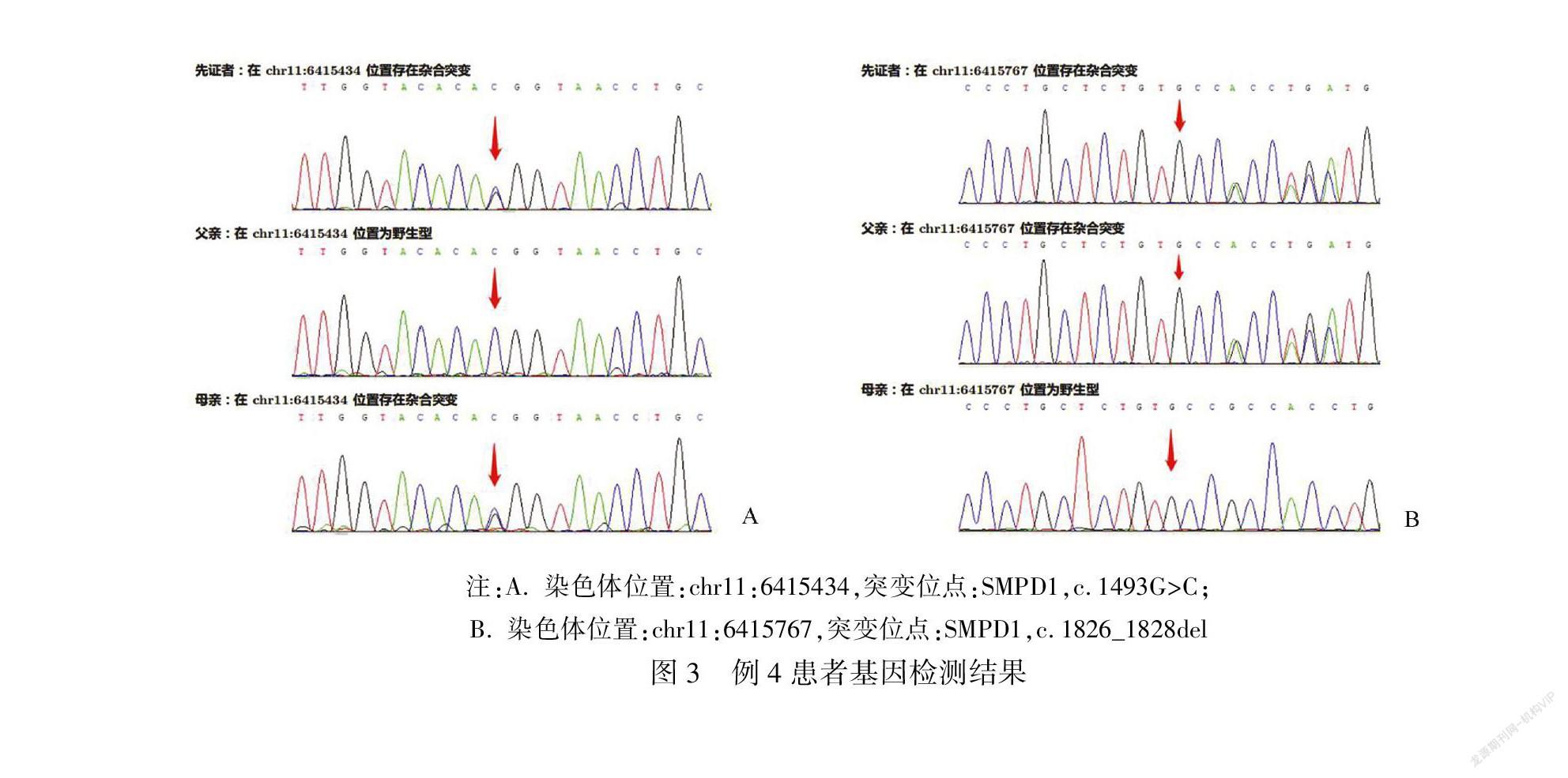
有1例患者(例4)行基因检测发现SMPD1基因上的两个突变c.1493G>C(p.R498P)、c.1826_1828del(p.609_610del)。见图3A、B。位点1突变的染色体位置为chr11:6415434,位点2突变的染色体位置为chrll:6415767,转录本NM_000543,位于第6个外显子。见表1。
3 讨 论
尼曼-匹克病B型是SMPD1基因突变所致,该基因定位于第11号染色体(11p15.4),含6个外显子,编码的蛋白是酸性鞘磷脂酶(ASM),共629个氨基酸,到目前为止,已经报道了170多种不同的导致疾病的SMPD1突变[2],包括错义突变、无义突变、缺失突变及剪接突变等,登记在HGMD中。ZAMPIERI等人[3]总结A/B型尼曼-匹克病人中报道的突变,发现其中错义突变占65.4%,移码突变占19%、无义突变占7%。而错义突变中70%都呈现出很低的ASM活性,其余30%活性较高的大部分来源于尼曼-匹克B型患者。SMPD1基因的热点突变在不同种群中存在很大的差异,这种基因突变上的种族差异性可能是导致尼曼-匹克病A型、B型在各种族中发病率存在极大差异的原因。国外研究[4]证实的尼曼-匹克病B型热点突变是△R608和G242R,北非的NPD-B型患者中,87%的突变类型为p.△R608,其他种族的患者中,大部分突变类型是罕见的,仅能在单个或数个家系中检测出。
本文例4患者检测到的SMPD1基因上的突变位点c.1493G>C(p.R498P)、c.1826_1828del(p.609_610del)为首次报道,c.1493G>C(p.R498P)在HGMD专业数据库和Clinvar数据库中无收录,但HGMD数据库中收录c.1493G>A(p.R498H)和c.1493G>T(p.R498L)为致病基因,两处位点报道的疾病均为尼曼-匹克病,SIFT预测、Polyphen2预测和MutationTaster预测为D(可能有害),参考美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)基因突变解读指南,该位点符合5条证据(PM1,PM PM5,PP3,PP4),考虑为可能致病。c.1826_1828del在HGMD数据库中无收录,Clinvar数据库中显示该位点对应的疾病和致病评级为:神经鞘磷脂/胆固醇沉积症(致病性),该位点的人群频率为0.000 在gnomAD数据库中东亚人群频率为0.00006,参考ACMG基因突变解读指南,该位点符合4条证据(PM PM3,PM4,PP4)考虑为可能致病。上方两个位点突变经家系验证为复合杂合突变,此复合杂合突变符合疾病隐性遗传模式,关联疾病与患儿临床表现吻合,此复合杂合突变可能导致疾病的发生。
B型NPD患者的常见临床表现包括肝脾肿大、脾功能亢进、肝功能损害、高脂血症和肺部受累,其他还可出现体格发育迟缓、眼底樱桃红斑、关节/肢体疼痛、挫伤、头痛、腹痛或腹泻等。国外关于NPD-B型患者10年的纵向研究[5]指出大约90%的患者以脾或肝脾肿大为主要症状,同时也是NPD-B型患者最常见的首发症状;而MCGOVERN等[6]的研究中也指出NPD-B型患者最常见的表现为脾大(78%)或肝大(73%),其他常见症状包括出血(49%)、肺部感染和呼吸急促(各占42%)、关节/四肢疼痛(39%),且患者脾脏的大小被證实与患者疾病的严重程度有关,包括可能会影响肝脏的功能、血脂的水平、身高、呼吸功能、造血功能,研究中患者中位死亡年龄为15.5岁,死亡原因包括肺炎、肝功能衰竭及出血,大多数患者在21岁前死亡,儿童病死率达19%。NPD-B型患儿中还常发现骨龄和青春期延迟,考虑与低胰岛素样生长因子1(IGF-1)水平有关,这类患者生长激素水平可能低于同龄儿童。
以肝脏肿大为首发表现的NPD患者,肝脏的组织病理学检查可以提供有关其结构方面的信息,以及肝细胞受损或纤维化的类型和严重程度,关系到治疗方案的选择。一篇关于17名成年人NPD-B型患者的研究[7]指出,几乎所有患者都存在不同程度的肝纤维化,其中2名患者进展为肝硬化,而他们没有任何肝功能衰竭的临床症状。本文有3例NPD-B型患者行肝脏穿刺检查,均发现泡沫细胞,病例1的肝组织显示肝小叶结构不清楚,肝细胞弥漫性肿胀;病例3的肝组织显示肝细胞索弥漫性疏松肿胀,肝窦狭窄,未见坏死,肝门管区旁可见泡沫细胞;病例4肝小叶结构存在,肝细胞体积明显增大,小叶内见点状坏死及小范围碎片状坏死,枯否氏细胞呈泡沫状。3例患者均有不同程度的慢性肝炎和纤维化表现,其中慢性肝炎为轻度,肝纤维化为中期F 与上述文献研究中肝组织病理学特征一致,但因病例数过少,不能确定肝纤维化程度是否与发病年龄相关,有待进一步研究。由于肝功能障碍和肝纤维化的程度可能是决定B型NPD患者预后的重要并发症,因此未来可针对基因型和表型与肝功能障碍、肝纤维化的相关性进行更深入的研究。
B型尼曼-匹克病的药物治疗目前尚无确定性,以对症及支持治疗为主,脾脏功能亢进时可行脾切除术。异基因造血干细胞移植也是治疗方法之一,能阻止内脏的进展及早期死亡[8]。一种称为olipudase alfa的人重组酸性神经鞘磷脂酶替代疗法有望治疗NPD-B型,动物试验中显示有效地提高了ASM酶的活性,减缓及逆转了对内脏器官的损害[9]。目前关于酶替代治疗在儿童和成人患者中分别进行Ⅱ期和Ⅱ/Ⅲ期的临床试验[10]。
总之,对于发现肝脾肿大、肝功能损害伴血脂异常的患者需考虑B型尼曼-匹克病的可能,并尽早完善基因检测,有助于早期诊断和治疗。
参 考 文 献
[1] SCHUCHMAN E H,DESNICK R J.Types A and B Niemann-Pick disease[J].Mol Genet Metab,2017,120(1/2):27-33.
[2] RHEIN C,MHLE C,KORNHUBER J,et al.Alleged detrimental mutations in the SMPD1 gene in patients with Niemann-Pick disease[J].Int J Mol Sci,2015,16(6):13649-13652.
[3] ZAMPIERI S,FILOCAMO M,PIANTA A,et al.SMPD1 mutation update:database and comprehensive analysis of published and novel variants[J].Hum Mutat,2016,37(2):139-147.
[4] KALMAN L,WILSON J A,BULLER A,et al.Development of genomic DNA reference materials for genetic testing of disorders common in people of Ashkenazi Jewish descent[J].J Mol Diagn,2009,11(6):530-536.
[5] WASSERSTEIN M P,DESNICK R J,SCHUCHMAN E H,et al.The natural history of type B Niemann-Pick disease:results from a 10-year longitudinal study[J].Pediatrics,2004,114(6):e672-e677.
[6] MCGOVERN M M,LIPPA N,BAGIELLA E,et al.Morbidity and mortality in type B Niemann-Pick disease[J].Genet Med,2013,15(8):618-623.
[7] THURBERG B L,WASSERSTEIN M P,SCHIANO T,et al.Liver and skin histopathology in adults with acid sphingomyelinase deficiency (Niemann-Pick disease type B)[J].Am J Surg Pathol,2012,36(8):1234-1246.
[8] 唐湘凤.尼曼-匹克病诊治进展[J].传染病信息,2019,32(2):154-157.
[9] WASSERSTEIN M P,JONES S A,SORAN H,et al.Successful within-patient dose escalation of olipudase Alfa in acid sphingomyelinase deficiency[J].Mol Genet Metab,2015,116(1/2):88-97.
[10] MCGOVERN M M,WASSERSTEIN M P,KIRMSE B,et al.Novel first-dose adverse drug reactions during a phase I trial of olipudase Alfa (recombinant human acid sphingomyelinase) in adults with Niemann-Pick disease type B (acid sphingomyelinase deficiency)[J].Genet Med,2016,18(1):34-40.
(收稿日期:2021-06-04 修回日期:2021-08-22)
(编辑:潘明志)
