NLRP3炎症小体参与下肢动脉腔内治疗后再狭窄发生的研究进展
2021-12-06刘宇婷杨硕菲薛冠华
刘宇婷,杨硕菲,薛冠华
(上海交通大学医学院附属仁济医院 血管外科,上海 200127)
随着社会老龄化的日益加剧和生活方式的改变,下肢动脉硬化闭塞症(arteriosclerosis obliterans,ASO)已成为仅次于冠心病和脑卒中的第三大高发的动脉粥样硬化(atherosclerosis,AS)性血管疾病,ASO在老年人群中的患病率超过10%[1-2]。随着腔内介入技术和器械的快速发展,血管腔内治疗成为ASO的一线治疗方式。但下肢动脉腔内治疗后再狭窄的发生率较高,影响了下肢动脉腔内治疗后的长期通畅。研究发现,金属裸支架置入后12个月再狭窄的发生率达30%,24个月再狭窄的发生率达50%[3]。目前,临床上尚无针对下肢动脉腔内治疗后再狭窄的有效干预措施,下肢动脉再狭窄的临床治疗已成为亟待解决的问题。再狭窄发生的病理基础是血管中膜的平滑肌细胞激活后迁移至内膜引起内膜过度增生,但具体发生机制尚不清楚。与冠状动脉和颅内动脉相比,下肢动脉长度长,运动频繁时易受到肌肉牵拉,使下肢动脉被动发生拉伸、压缩和扭转等形态学变化。下肢动脉再狭窄既受内膜过度增生的影响,又合并动脉形变引起的支架断裂和支架变形等物理因素的作用[4]。下肢动脉腔内治疗后再狭窄发生的具体机制需要深入研究。
血管平滑肌细胞(vascular smooth muscle cell,VSMC)在正常生理状态下处于收缩型,在血管损伤或受炎症刺激后,可从静止的收缩型转变为主动的合成型,从动脉中膜迁移到内膜并参与内膜增生,VSMC表型转化促进炎症反应和动脉粥样硬化形成,也与腔内治疗后再狭窄的发生密切相关[5-6]。同时,动脉的拉伸和压缩变形可调节VSMC增殖、凋亡、表型转化、迁移、排列并刺激细胞外基质重塑[7]。研究发现,局部炎症反应参与VSMC的表型转化和细胞外基质的重塑,炎症小体的激活在此过程中扮演重要角色。NOD样受体家族含pyrin结构域蛋白3(NOD-like receptor family pyrin domain-containing protein 3,NLRP3)炎症小体参与众多心血管疾病的发生和发展,在腔内治疗后再狭窄中也发挥重要作用[8-9]。
1 NLRP3炎症小体的结构和功能
1.1 NLRP3炎症小体的激活与NLRP3/IL-1β通路炎症小体于2002年首次被发现,它是由多种蛋白质组成的胞质内蛋白复合体,在固有免疫和炎症相关疾病的发生过程中发挥重要作用[10]。炎症小体的过度活化参与动脉粥样硬化、糖尿病(diabetes mellitus,DM)以及恶性肿瘤等多种疾病的发生[11]。炎症小体包括NLRP3、NLRP1、AIM2、NLRC4和pyrin等多种亚型,目前对NLRP3炎症小体的研究最为深入[12]。NLRP3炎症小体是由结合分子(NLRP3)、衔接分子(ASC,包含CARD结构域的凋亡相关斑点样蛋白)和效应分子(Caspase-1)组成的多蛋白复合体[13]。NLRP3炎症小体激活通过两步信号实现:第一信号是启动信号,由病原体相关分子模式(TLR)或危险相关分子模式诱导,TLR磷酸化后激活NF-κB,在细胞核中,NF-κB促进NLRP3、IL-1β前体(pro-interleukin-1β,pro-IL-1β)和IL-18前体(pro-interleukin-1β,pro-IL-18)转录,翻译后它们以非活性形式留在细胞质中[13];第二信号是激活信号,由多种结构不同的激动剂触发,包括环境晶体污染物(如二氧化硅)、病原体衍生的配体(如成孔毒素和核酸)以及内源性危险信号(如血清淀粉样蛋白A和三磷酸腺苷)。NLRP3炎症小体组装成复合体后通过效应蛋白Caspase-1进行自我切割和活化,活化的Caspase-1可对pro-IL-1β和pro-IL-18进行切割,形成有活性的IL-1β和IL-18并释放到细胞外,募集炎症细胞聚集,增加炎症反应,活化的Caspase-1还能够切割Gasdermin D的N端和C端,诱导细胞膜穿孔,从而导致细胞焦亡[14-15]。离子交换、溶酶体破坏、活性氧(reactive oxygen species,ROS)产生、线粒体功能障碍、代谢变化和内质网应激等均可导致NLRP3炎症小体激活[16]。
1.2 NLRP3炎症小体在VSMC中的作用VSMC是构成动脉壁的主要细胞之一,α平滑肌肌动蛋白(α-smooth muscle actin,α-SMA)是VSMC的骨架蛋白,通常可作为VSMC的蛋白标志物。体外试验诱导的钙化VSMC和钙化的股动脉临床样本中NLRP3炎症小体复合物表达上调,IL-1β分泌增加,沉默NLRP3蛋白可抑制VSMC钙化[17]。高糖处理后的VSMC可检测到NLRP3炎症小体表达上调,释放大量IL-1β且加速VSMC钙化,抑制NLRP3炎症小体表达对VSMC钙化有拮抗作用[18]。氧化三甲胺(trimethylamineN-oxide,TMAO)通过激活NLRP3炎症小体和NF-κB促进血管钙化,抑制NLRP3炎症小体和NF-κB可以减弱VSMC的钙化[19]。在自发性高血压大鼠(spontaneously hypertensive rats,SHR)模型中,主动脉中膜VSMC中NLRP3炎症小体被激活且IL-1β的表达水平上调;沉默NLRP3基因对SHR有降压作用,抑制SHR主动脉中VSMC表型转化和增殖,调控血管重塑过程[20-21]。NLRP3炎症小体激活不但促进炎症反应的发生,还促进VSMC转变为肌源性泡沫细胞[22]。小鼠和人动脉粥样硬化斑块检测结果显示,Caspase-1和IL-1β与α-SMA在斑块内分布区域基本重合[23]。尿毒症小鼠和尿毒症患者的动、静脉瘘内膜病变中NLRP3表达上调,特异性NLRP3基因敲除小鼠VSMC的增殖、迁移、表型转化等能力均减弱[24]。慢性间断缺氧诱导肺和心脏CD11b+细胞超氧化物歧化酶2表达降低,CD11b+细胞中NLPR3炎症小体被激活,IL-1β、IL-18分泌增加,肺VSMC异常增殖导致肺动脉高压[25]。一方面VSMC中NLRP3炎症小体被激活,释放IL-1β等炎性因子促进血管钙化,另一方面由于NLRP3在免疫细胞中高表达,释放大量IL-1β等炎性因子,促进VSMC增殖和表型转化。以上研究结果均表明,NLRP3炎症小体过度激活在血管性疾病VSMC中起着直接或间接作用。
2 NLRP3炎症小体在下肢AS和再狭窄中的作用
AS是一种全身性疾病,ASO是下肢动脉因粥样斑块导致血管狭窄甚至闭塞,是全身AS在下肢的表现[26]。AS也是一种慢性炎症性疾病,以VSMC增殖和迁移、白细胞浸润、内皮功能障碍和长期脂质积聚为主要特征[6]。已有研究证明,NLRP3炎症小体激活后分泌的IL-1β能够促进VSMC增殖,增加白细胞募集和黏附,上调多种促炎因子表达[27]。IL-18和IL-1β是NLRP3炎症小体调控的关键细胞因子,它们在AS斑块组织中高表达,NLRP3炎症小体的激活参与了AS的发生和发展[28]。NLRP3炎症小体可以被多种内源性危险信号激活,这些危险信号在AS病变中大量存在。Duewell等[29]研究发现,高脂饮食诱导的AS早期病变中存在微小的结晶胆固醇,可以激活NLRP3炎症小体,促进IL-1家族细胞因子的成熟和分泌,NLRP3炎症小体在早期AS中起促进作用。进一步研究发现,NLRP3炎症小体不但能识别致病物质,短期内释放大量的炎症信号,还能够感知高脂高糖饮食进一步放大炎症反应[30]。NLRP3在ASO病变组织的动脉中膜和内膜中高表达,在正常对照组中低表达[22]。颈动脉AS斑块中NLRP3、ASC、Caspase-1、IL-1β和IL-18表达显著上调[31]。研究指出,随着粥样斑块的增大,NLRP3的表达也增加,抑制NLRP3和Caspase-1可阻止AS斑块相关组织和细胞释放IL-1β,抗IL-1β治疗可缓解AS的发生[32-33]。MCC950是NLRP3炎症小体的抑制剂,可降低体内IL-1β的产生,用于治疗NLRP3相关的自身免疫和炎症性疾病[34]。能否使用MCC950抑制NLRP3炎症小体激活从而减缓下肢动脉AS的发生、发展尚需要进一步研究。
内膜增生是动脉腔内治疗后再狭窄的病理学特征之一,涉及VSMC异常增殖和迁移以及细胞外基质聚积,最终导致管腔狭窄。血管腔内治疗后引发一系列的炎症反应,炎性细胞被激活后分泌炎症介质和生长因子,使VSMC异常增殖并逐渐从中膜向内膜移动,并发生表型转化,由收缩型转化变为合成型,分泌胶原成分参与内膜增生[35]。大鼠颈动脉新生内膜组织中NLRP3、Caspase-1和IL-1β表达水平明显上调且血清中IL-1β和IL-18水平显著升高[36]。再狭窄小鼠模型内膜增生主要由大量的VSMC组成,病变组织中NLRP3和Caspase-1强烈表达,NLRP3基因敲除小鼠与野生型小鼠对比内膜增生明显减少[37]。再狭窄明确的病理学机制是内膜增生,最近研究发现新生AS也是再狭窄病变的一种机制[38]。早期(<1年)和晚期(>1年)再狭窄的组织特征存在差异,前者主要由内膜增生介导,而后者以新生AS为主要机制[39]。血管腔内治疗后局部炎症反应促进VSMC增殖、迁移和细胞外基质沉积,造成内膜不断增厚。在再狭窄小鼠模型中,股动脉损伤部位和损伤后新生内膜病变中观察到ASC表达增强,而ASC基因敲除小鼠内膜形成减少且病变中IL-1β和IL-18表达显著降低[40]。颈动脉再狭窄小鼠模型中VSMC细胞内NLRP3、ASC与α-SMA共定位表达,表达量显著上调,炎症小体抑制剂MCC950和WEHD能够通过阻断Caspase-1和IL-1β的表达和分泌阻止VSMC的过度增殖和迁移[9]。秋水仙碱和甲氨蝶呤可分别抑制NLRP3炎症小体激活后产生的IL-1β和NLRP3炎症小体激活物NF-κB,有效抑制AS的发生[41]。这些药物能否通过抑制新生AS从而抑制腔内治疗后再狭窄发生还需要进一步实验证实。
3 结语
综上所述,腔内治疗后再狭窄具有两种病理机制,一是内膜增生,二是新生AS。腔内治疗导致血管炎症反应,促使VSMC异常增殖并逐渐从中膜向内膜移动且发生表型转化,细胞外基质不断沉积,造成内膜增生。VSMC胞核中NF-κB促进NLRP3、pro-IL-1β转录与翻译后以非活性形式留在细胞质中,NLRP3、ASC和Caspase-1聚集促使NLRP3炎症小体组装和激活。活化的Caspase-1对pro-IL-1β进行加工,从而促进IL-1β的成熟和分泌,IL-1β介导炎症反应且促进VSMC增殖。NLRP3炎症小体激活促进VSMC转变为肌源性泡沫细胞,参与新生AS形成(图1)。
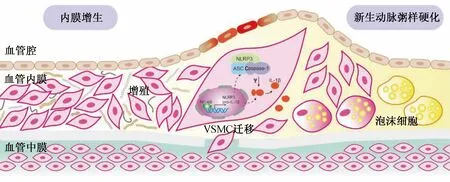
图1 腔内治疗后再狭窄的病理机制示意图
下肢动脉腔内治疗后再狭窄的治疗仍是当前血管外科医生面对的重要挑战。当前研究显示,NLRP3炎症小体的激活可促进VSMC表型转化和迁移,导致血管内膜增生,引起再狭窄的发生。对于NLRP3炎症小体的深入研究可为阐明下肢动脉腔内治疗后再狭窄发生机制提供全新的思路。对NLRP3炎症小体及其相关分子通路的干预可能阻止血管内膜的过度增生,减少再狭窄的发生,有望为再狭窄的治疗提供新的突破口。目前,下肢腔内治疗中载药器械所载药物主要为紫杉醇,紫杉醇涂药球囊和涂药支架能够显著减少腔内治疗后再狭窄的发生[42]。随着紫杉醇涂药器械的广泛应用,有研究人员发现,紫杉醇作为一种常用的肿瘤化疗药物存在增加患者全因死亡率的风险,引起了巨大争议和讨论[43]。未来针对NLRP3炎症小体为靶点的相关药物进行新一代载药器械的研发有望在减少再狭窄发生的同时避免增加死亡风险的可能。
