不同阳离子分子筛中CO2吸附结构形成与变化机理
2021-12-02杨凯中吴静怡
杨凯中,杨 光,吴静怡
(上海交通大学 制冷与低温工程研究所,上海 200240)
0 引言
分子筛在空气分离系统[1-3]、低温真空绝热储存系统[4-5]等低温与真空领域有重要应用。在空气分离系统中,空气通过低温精馏进行分离之前,须使用分子筛去除空气中CO2和水分等杂质,防止其在精馏过程中冻结堵塞精馏塔。在LNG储罐车中,分子筛常用于吸附绝热夹层中释放出来的气体,从而维持夹层的高真空度,实现较好的绝热效果。由于空气中CO2浓度低,且绝热夹层总压较低,因此在较低的CO2分压下发生吸附。所以吸附材料的低压吸附性能尤其重要[6-7]。
空分纯化系统大多采用低压CO2吸附性能较好的X型分子筛去除空气中的CO2。而随着空分设备朝大型化发展以及空气中CO2浓度的增加,开发吸附量更大的CO2吸附剂,对于延长吸附切换周期,降低空分能耗具有重要意义[7]。由于X型分子筛成本较低,利用改性X分子筛提升CO2吸附量具有较好的经济性。X分子筛的改性主要有两种途径:一是改变分子筛硅铝比;二是改变分子筛阳离子种类。其中,低硅铝比X分子筛(LSX)具有更好的低压CO2吸附性能,由此被应用在一些新的空分项目中[8-9]。作者前期研究了降低硅铝比来强化分子筛吸附的机理[10],发现其较多的高能量吸附位点的数目是CO2吸附强化的主要原因。低硅铝比分子筛中高能位点数目增加是由其阳离子浓度的增加导致的,说明了阳离子在分子筛吸附CO2过程中具有重要作用。因此,在另一方面,也有通过改变阳离子的方法强化CO2吸附能力的研究。但这些研究往往停留在指出不同阳离子分子筛吸附能力的宏观差别[3]的层面上,或者利用不同阳离子的性质差异对吸附能力进行宏观解释[11-12],缺乏对阳离子影响吸附能力的微观机理的解释。
作者在之前关于分子筛吸附位点特征的分子模拟研究中发现[10,13],从微观角度看,吸附位点特征变化较好地解释了低硅铝比分子筛的吸附强化机理。目前鲜有将此方法应用于揭示不同阳离子分子筛的CO2吸附机理的研究。吸附结构的特征对CO2的吸附能力有很大影响,反映了CO2的吸附机理。因此,不同阳离子分子筛中吸附结构的理论与模拟研究,对于从微观层面揭示不同阳离子分子筛中CO2吸附强化机理,指导开发具有高CO2吸附性能的吸附剂材料的研究具有重要意义。
本文首先基于原子相互作用基本理论,将阐述CO2吸附在分子筛时所形成的吸附结构受阳离子半径或极化率的影响,并由此得到LiX、NaX和KX三种不同阳离子分子筛中吸附结构的变化规律。随后对不同阳离子分子筛中的吸附结构及其CO2吸附性能进行分子模拟,验证上述分析结论的准确性。
1 方法
1.1 模型分子结构
X分子筛是一种八面沸石分子筛(FAU),化学式为 M86Al86Si106O384,其中 M 表示 Li+、Na+、K+等阳离子。X分子筛骨架中,硅氧(或铝氧)四面体相互连接组成钠笼(β笼),β笼再通过六元环相互连接形成超笼(α笼),其结构示意图如图1所示(由VESTA绘制[14])。CO2在分子筛中一般通过非键结作用吸附在X分子筛中的α笼中。当吸附量不是很大时,CO2一般吸附在α笼中靠近分子筛骨架的位置,其在α笼中的典型吸附位置如图1所示。CO2不能进入孔径较小的β笼,因此β笼被排除在模拟吸附区域外。在模拟中,分子筛骨架原子位置固定[15],骨架的硅铝分布是在满足路易斯准则的前提下[16],通过随机替换Al原子获得,这种随机替换不会对模拟结果产生较大影响[17-18]。CO2分子中C=O键长为0.114 9 nm,O=C=O的角度为179.997°[19]。如图1所示,分子筛中的86个阳离子中,64个阳离子分布在32个SI′位置和32个SII位置[20],并在模拟中位置固定。剩余22个阳离子不能确定其具体位于哪一个SIII或SIII′位置,设置其在模拟中可移动,这样的方式能够有效平衡模型的准确性与计算的经济性,其可靠性在前期研究中已说明[13]。
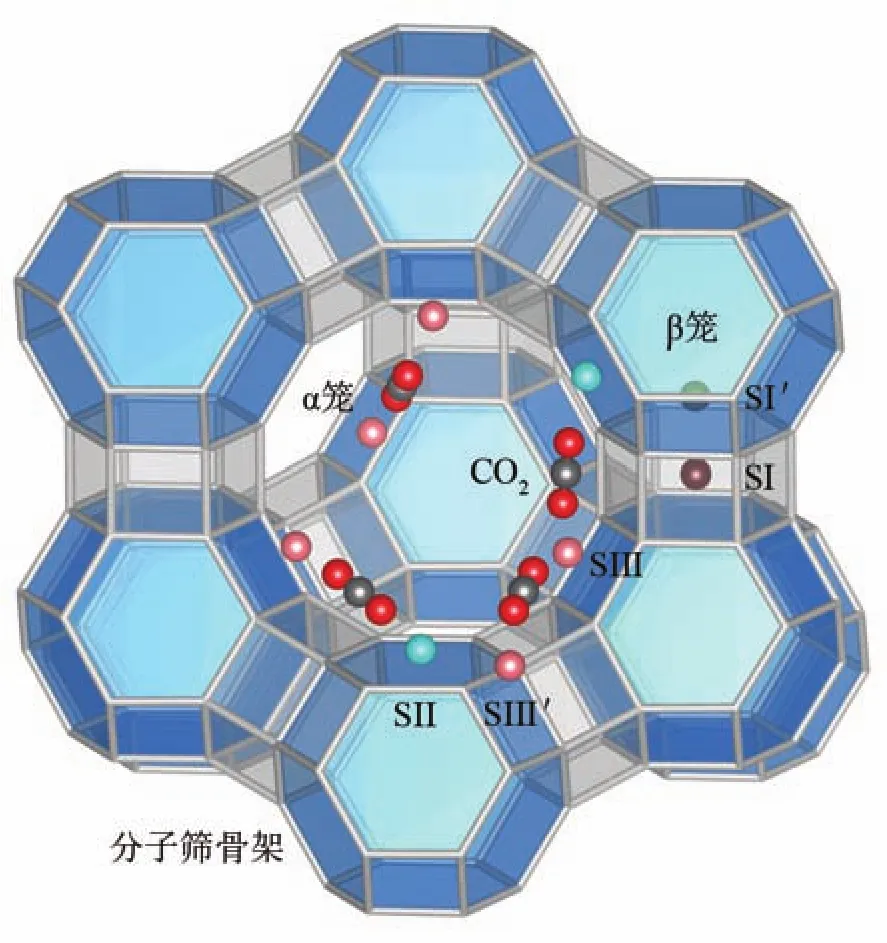
图1 八面沸石分子筛结构及阳离子与CO2位置示意图Fig.1 Schematic view of faujasite and position of cations and CO2molecules
1.2 模拟细节
本文研究的吸附过程中的主客体相互作用是指代表主体的分子筛与代表客体的CO2分子间的相互作用。吸附结构能量主要由两部分构成:一部分是主客体相互作用的范德华势能Evdw;另一部分为主客体相互作用的电势能Ecoul。i原子和j原子的Evdw和Ecoul的计算表达式分别为:
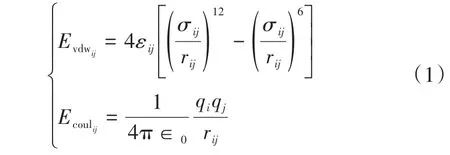
式中:εij和σij为相互作用对参数,分别反映了相互作用的最小势能大小和势能为0的位置;q为原子的电荷;rij为原子间的相互距离。
对于吸附结构的分子模拟采用正则系综的蒙特卡洛(CMC)模拟方法,模拟过程中的最低能量构型将会被记录并用于分析吸附结构。对于吸附等温线的模拟则采用巨正则系综的蒙特卡洛模拟(GCMC)方法。对于NaX分子筛,模拟力场采用Fang等[21]报道的基于第一性原理推导的力场,相比于经验或半经验力场,其更适合揭示微观的物理机制[22]。该力场的可靠性已由其作者[21,23]和其他研究者验证[24]。
分子模拟采用RASPA 2.0软件[25],Evdw截断半径为1.2 nm,Ecoul采取Eward求和方式求解。在每个算例中均使用1×105个MC循环步作为插入CO2的生产步,另外1×105个MC循环步作为CO2的平衡步。MC循环步是RASPA中的定义,1个MC循环步中会对系统中每个可活动的分子进行一次操作(包括平移、旋转、替换等),在之前研究中,已经证明该步长能给出较精确的模拟结果[26]。
2 结果与讨论
CO2吸附在分子筛中特定位点并与阳离子及分子筛骨架形成的结构被称为吸附结构。这种结构具备一些典型特征,例如过往文献认为Na+与CO2会形成近似线性的Na+…O=C=O结构,且Na离子的数目和位点类型会影响该吸附位点的强度(位点结合能)[21,27]。前期研究[13]发现,该吸附结构的另一典型特征是分子筛骨架中的O原子(Oz)会与CO2分子形成垂直结构,并指出通过对单原子与CO2相互作用能分析,可以给出吸附结构特征的形成原因。吸附结构的特征能对CO2的吸附能力有很大影响,反映了CO2的吸附机理。本节将讨论不同阳离子中的吸附结构变化。
2.1 不同阳离子分子筛吸附构型的理论研究
阳离子与单个CO2相互作用的最小势能点可由式(1)计算,反映了相互作用的最稳定状态,因此能在某种程度上体现吸附结构的特征。式中σ参数表示两原子间相互作用势能为0时的原子间距,ε表示相互作用的最小势能。由于范德华力本质上是原子的电子云之间的相互作用引起的,因此σ和ε参数受离子半径大小和极化率影响。对于碱金属X分子筛,Maurin等[11]提出式(2)来衡量该影响:

式中:R和α分别为离子半径与极化率。M1和M2代表不同阳离子。阳离子与其他原子相互作用的混合规则均采取几何平均:

M1和某一原子的相互作用参数σM10和εM10分别为:
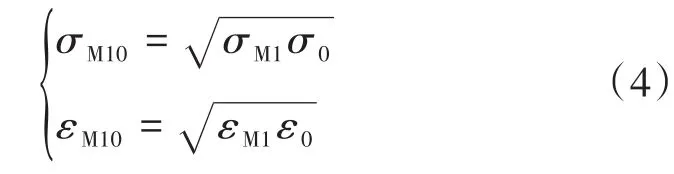
M2和该原子的相互作用参数σM20和εM20与σM10和εM10关系为:

根据式(4)可以计算不同的阳离子半径以及极化率对力场参数的影响,从而根据式(1)计算出理想情况下阳离子与CO2吸附的相互作用势能。图2是当离子半径为0.116 nm,极化率为α=1.80×10-3nm3(对应于Na+)时,阳离子与CO2相互作用的等势线。可知其相互作用能的最小值点位于CO2分子所在直线上,且最小点距离O原子距离(M-O)约为0.241 nm。

图2 离子R=0.116 nm、α=1.80×10-3nm3时Na+与CO2相互作用等势能线图Fig.2 Isopotential curve of cation-CO2interaction potential energy at R=0.116 nm,α =1.80×10-3nm3
当离子半径变化时,相互作用参数也会变化。由于M-CO2相互作用的最小值始终在CO2分子所在直线上[13],可通过M在CO2分子所在直线移动时MCO2相互作用能量分布变化研究最小值点随离子半径的变化规律。图3(a)给出了阳离子半径分别为0.073、0.116、0.152 nm时CO2分子所在直线上的相互作用能量分布曲线。可以看出,随着离子半径的增加,离子与CO2相互作用最小值点到C原子的距离不断增加,且相互作用的能量最小值也不断减小。图3(b)中极化率的变化对能量最小值点也有类似的影响,即极化率增加,离子与CO2相互作用最小值点到C原子的距离增加,最小值点能量减小,但极化率对能量最小值点的影响相对较小。
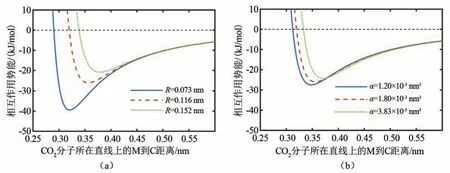
图3 不同阳离子半径和极化率下CO2分子所在直线上M-CO2相互作用势能曲线Fig.3 M-CO2interaction potential energy curve on the line of CO2molecule with different cation radius and polarizability
单个阳离子与CO2相互作用的能量最小值点在一定程度上可以反映在实际吸附结构中的位置与能量。图4显示了不同阳离子半径及极化率对M-CO2的吸附结构中M-O距离及与其能量的影响。图中可以看出,在一定范围内,离子半径的增加会增大形成M-CO2的吸附结构中M-O距离,降低M-CO2的能量。极化率对吸附结构有相似的影响,但对吸附结构的影响小于离子半径。影响规律可以由下述机制解释:当极化率增加、离子半径变大时,在离子周围受该离子排斥力影响的区域也变大,使得其他原子与该离子相互作用时平衡距离增大,从而使其相互作用的能量减小。
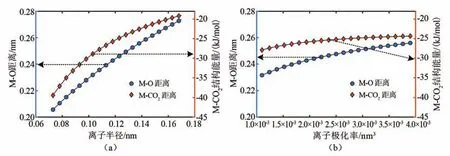
图4 阳离子半径和极化率对能量最小值及其位置影响Fig.4 Influence of cation radius and polarizability on the minimum energy and position of M-CO2interaction structure
文献[21]给出了Na+与CO2的作用参数(σM10和εM10),则由式(4)及常见的Li+、Na+和K+离子半径与极化率数据如表1所列[11],可以计算不同阳离子与CO2相互作用参数,从而得到LiX、NaX和KX中吸附结构的能量和结构变化关系,如图5所示。可以看出,当分子筛中的阳离子按Li+、Na+、K+的顺序变化时,其形成M-CO2的最小能量构型的能量是逐渐减小的,且M-O的距离也是逐渐减小的。产生这种现象的原因是从Li+-Na+-K+,阳离子的半径和极化率增加。
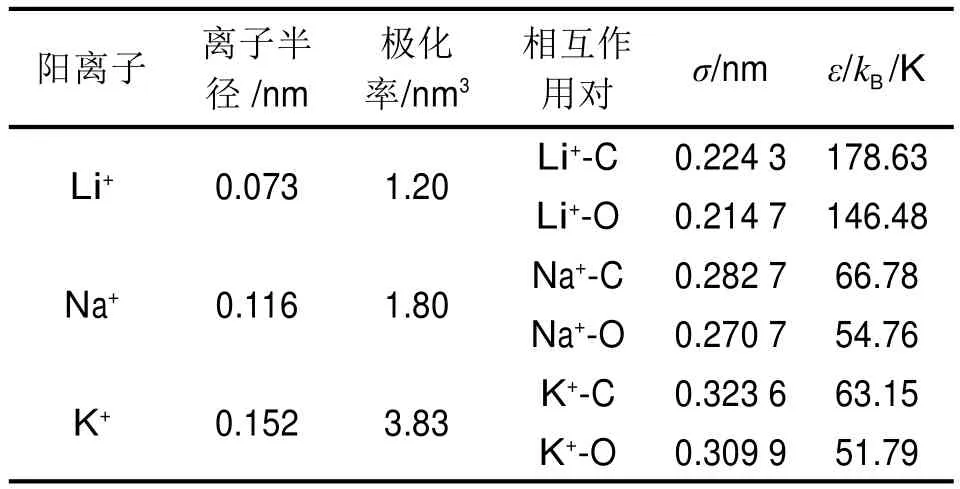
表1 不同阳离子离子半径、极化率,以及与CO2相互作用力场参数Tab.1 The radius,polarizability,and force field parameters of cations

图5 不同阳离子类型分子筛中M-CO2最小能量构型的能量及位置变化Fig.5 Minimum energy and position of M-CO2interaction structure in zeolites with different cations
2.2 不同阳离子分子筛中吸附构型的分子模拟研究
从理论上研究了理想情况下阳离子对吸附结构的影响机理,通过简洁且具有明确物理意义的模型给出不同阳离子影响所形成的吸附结构的机理,其结论具有一般性。而分子模拟可以模拟真实的不同阳离子分子筛中CO2的吸附结构。一方面,分子模拟可以验证理论研究中一般性结论在实际运用中的可靠性;另一方面,理论研究可以为分子模拟中获得的现象提供机理层面的解释。因此,采取分子模拟的方法模拟实际情况下CO2在不同阳离子分子筛中的吸附结构。分子模拟中Li+和K+与CO2分子的相互作用力场参数如表1所列,其他参数假设与原力场相同。图6给出了分子筛晶胞只吸附一个CO2分子时,不同阳离子分子筛中吸附结构的模拟结果。

图6 不同类型分子筛α笼中的CO2吸附结构模拟快照图Fig.6 Snapshot of CO2 adsorption structure in α-cage of different kinds of zeolites
由图6可知,在LiX、NaX和KX所形成的不同吸附结构中,阳离子至O的最小距离分别为2.45、2.52和0.303 nm。即LiX中M-O的距离最小,而KX中M-O的距离最大,这和上述理论分析结论相符。LiX、NaX及KX中吸附结构的能量分别为-57.9、-53.1、-41.3 kJ/mol,也符合图5的变化规律。此外,LiX和NaX中吸附结构的M-O距离差异较小,对应的能量差异也较小。这与图4中M-CO2吸附结构的能量与M-O的距离成反比的结论也相符。同时,需要注意的是,在实际的相互作用中,CO2在分子筛中形成的吸附结构中并不是严格的M与CO2成直线、Oz与CO2相垂直的关系,但总体上M…CO2⊥Oz的结构特征得以保留。此差异是由实际情况中更复杂的相互作用引起的,在之前的研究中,作者也证明了若考虑M与CO2和Oz的共同作用,M…CO2的线性结构会发生偏转[13]。此外,还注意到CO2会同时与两个阳离子作用,形成M-CO2-M的双阳离子的吸附结构,这与两个M(位于SIII或SIII′)的间距较小有关。
图7给出了通过分子模拟计算的不同阳离子与CO2中O原子的径向分布函数(RDF)。从图中可以看出,LiX、NaX、KX中RDF的第一个峰值分别为0.205、0.248、0.289 nm。由于RDF描述了O原子周围阳离子的密度分布,因此上述距离代表Li+、Na+、K+最有可能出现在距离CO2分子中距O原子0.205、0.248、0.289 nm的位置,将上述峰值位置绘制于图5中可以发现,不同阳离子的RDF峰值位置与图5中单个阳离子与CO2分子相互作用势能最小时M-O距离(0.200、0.241、0.281 nm)基本相符,这也说明了上节中的最小势能点的理论分析在一定程度上反映真实的吸附结构情况。

图7 不同类型分子筛中M-O距离的径向分布函数Fig.7 Radial distribution function of M-O distance in different kinds of zeolites
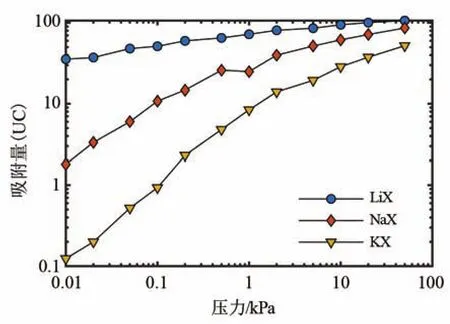
为排除由单个模拟结果偶然性的影响,进一步验证LiX、NaX和KX中吸附结构变化规律,使用CMC模拟了不同吸附量下不同阳离子分子筛中的吸附结构,并计算了吸附结构中的平均M-O距离。如图8所示,在吸附量为0~100个每晶胞(UC)的范围内,吸附结构中M-O的平均距离基本满足LiX 通过对实际情况中CO2在不同分子筛中的分子模拟可知,吸附结构的最小能量构型中M-O距离(图6、图8)以及M-O的径向分布函数最大峰值位置(图7),均与上节中通过单阳离子与CO2相互作用最小势能结构中的M-O距离的变化趋势(图5)相同。这说明,单个阳离子与CO2相互作用反映出的吸附结构特征的变化规律,能够一定程度上反映在真实情况下考虑了多个阳离子之间、多个CO2分子之间、多个吸附结构之间相互作用时的吸附结构的特征。 图8 不同阳离子分子筛在不同吸附量下吸附结构中的M-O平均距离Fig.8 Average M-O distance in adsorption structure in different zeolites under different loading 利用GCMC模拟不同阳离子的分子筛的CO2吸附等温线,如图9所示。可以看出CO2吸附量LiX>NaX>KX,这与之前的文献结论一致[3]。此现象可从吸附动力学的角度解释:当CO2吸附在较大能量的吸附位点时,其吸附停留时间更长,因此吸附与脱附达到平衡时的吸附量更大。此外,LiX中M-O距离较小,能容纳更多的CO2形成的吸附结构也是LiX具有较大饱和CO2吸附量的一个原因。 图9 不同阳离子分子筛在303 K下的CO2吸附等温线Fig.9 CO2adsorption isotherm on zeolites at 303 K with different cations 综上所述,LiX相比与NaX具有更强的CO2吸附性能。其机理主要是Li具有较小的阳离子半径和极化率,能在分子筛吸附时与CO2形成更紧凑、能量更大的吸附结构。因此,LiX能容纳数目更多、吸附结构更稳定的CO2分子。 通过理论分析与分子模拟相结合的方法,研究了不同阳离子分子筛中所形成的M-CO2吸附结构变化机理。分子筛吸附结构的能量以及其中M-O的距离与阳离子的半径以及极化率有关:阳离子半径越大、极化率越强时,M-O的距离越大,吸附结构的能量(绝对值)越小。在LiX、NaX和KX中,单个M-CO2吸附结构中M-O距离的理论计算值分别为 0.200、0.241、0.281 nm。而 LiX、NaX 和 KX 的M-O的径向分布函数的分子模拟显示M-O距离分别为0.205、0.248、0.289 nm。分子模拟与理论计算吻合较好,说明了吸附结构的变化可由单个阳离子与CO2相互作用的最小势能点的变化来反映。此外,分子模拟结果显示,单位吸附量下,LiX、NaX和KX分子筛中的M-CO2吸附结构中的M-O距离分别为0.245、0.252、0.303 nm,其能量分别为-57.9、-53.1、-41.3 kJ/mol。在不同吸附量下,CO2吸附结构中M-O距离也满足LiX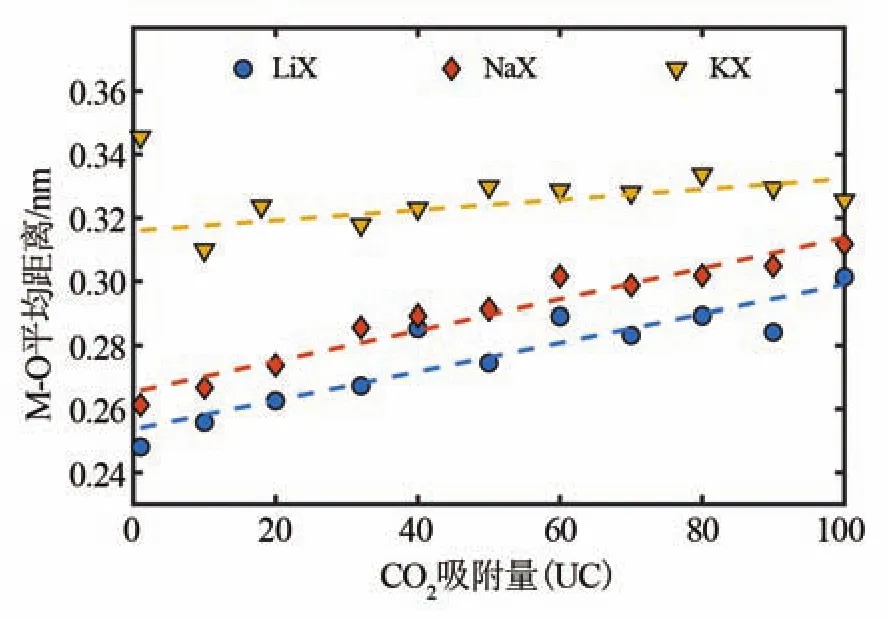
3 结论
