高效液相色谱-串联质谱法测定龙血碣药材中苏丹红Ⅳ
2021-12-02李柯杨蕾孟令嘉
李柯,杨蕾,孟令嘉
(首都医科大学附属北京世纪坛医院,北京 100038)
龙血竭为百合科植物剑叶龙血树[Dracaena cochinchinensis(Lour.)S.C.Chen]的含脂木材经提取得到的树脂,主要产于东南亚国家和我国云南等地,具有活血散瘀、止血止痛、敛疮生肌的作用,主要用于外伤出血,跌打损伤等症状[1-3]。目前对龙血竭药材的研究主要有龙血竭的化学成分[4]、高效液相色谱指纹图谱[5]、有效成分含量测定[6-8]及部分非法染色[9]。文献报道称有部分不法商家为了商品的外观色泽鲜亮,在龙血竭中掺杂非法染色剂,对人们的用药安全产生一定的危害,文献报道的龙血竭药材中主要检出苏丹红系列和808猩红等染料[9],苏丹红系列是一种人工合成的偶氮型燃料[10-12],其代谢产物产生的苯胺和萘胺具有较强的致癌作用,欧美国家已经明确禁止将苏丹红添加至食品与药品中。目前对苏丹红的研究主要为食品中非法添加的检测[13],苏丹红的测定方法主要有薄层色谱法[14]、紫外分光光度法[15]、液相色谱法[16]、气相色谱串联质谱法[17]和液相色谱串联质谱法[18-19]等。笔者参考文献资料和中国药典[20-21]对10批不同来源的龙血竭药材非法染色进行研究,通过紫外光谱、液相色谱串联质谱进行鉴别和确认,比目前现有文献报道方法更全面,从定性到到定量,并对检出的苏丹红Ⅳ进行含量测定的方法学验证,为龙血竭药材中非法染色的鉴别和苏丹红Ⅳ的含量测定提供实验依据,进而可以更加全面的评价龙血竭药材的质量,从而保证其临床用药的安全性和有效性。
1 实验部分
1.1 主要仪器与试剂
高效液相串联质谱仪:Agilent1260-G6410型,MassHunter采集软件,包括四元洗脱泵,DAD检测器,美国安捷伦仪器公司。
双频数控超声波清洗器:KQ-700VDB型,江苏昆山超声仪器公司。
电子天平:BSA-124S型,感量为0.1 mg,德国赛多利斯公司。
苏丹红Ⅳ:纯度(质量分数)为94.0%,批号为40121,德国Dr Ehrenstorfer GmbH公司。
乙腈:色谱级,美国Fisher公司。
氮气:纯度(体积分数)不小于99.999%,北京天昊气体有限公司。
实验用水为超纯水。
实验所用其它试剂均为分析纯。
龙血碣药材分别来源于云南西双版纳、广西桂林、贵州铜仁、缅甸、泰国等东南亚地区,所有药材购自河北安国中药材市场,药材信息见表1。

表1 药材的样品信息
1.2 仪器工作条件
1.2.1 色谱
Agilent-C18XDB色 谱 柱(250 mm×4.6 mm,5 μm,美国安捷伦科技有限公司),流动相:乙腈-0.02%甲酸溶液(体积比为95∶5),流量为1.0 mL/min;柱温:30 ℃;检测波长:518 nm;进样体积:10 μL;理论塔板数苏丹红Ⅳ峰计算不低于15 000。苏丹红Ⅳ对照品和伪品龙血竭以及龙血竭药材的色谱图分别见图1、图2、图3。

图1 苏丹红Ⅳ对照品色谱图

图2 伪品龙血竭色谱图
图3 龙血竭药材色谱图
1.2.2 质谱
采用三重四级杆质谱仪进行测定;ESI离子源;正离子模式扫描;干燥气和雾化气:氮气,压力为137.9 KPa(20 psi),流量为 3.0 mL/min;加热气:空气;干燥气体温度:300℃;毛细管电压:3.5 kV;质量扫描范围:50~500 u;多反应监测(MRM)模式;碰撞能量:30 eV;裂解电压:110 V;苏丹红Ⅳ的一级质谱和二级质谱图见图4、图5。

图4 苏丹红Ⅳ对照品的一级质谱和二级质谱碎片图

图5 伪品龙血竭样品的一级质谱和二级质谱碎片图
1.3 溶液的配制
1.3.1 对照品溶液的制备
精密称取苏丹红Ⅳ对照品7.50 mg置于50 mL量瓶中,加乙腈溶解定容至标线,摇匀,作为对照品储备溶液。精密吸取上述苏丹红Ⅳ对照品储备液5 mL置于25 mL量瓶中,加乙腈稀释至标线,摇匀,配制成苏丹红Ⅳ质量浓度为30 μg/mL的对照品溶液。
1.3.2 供试品溶液的制备
精密称取上述批号200101的疑似染色龙血竭药材粉末0.2 g,加乙腈25 mL,超声处理10 min,静置,放凉,采用0.22 μm的微孔滤膜滤过,取续滤液作为供试品溶液。
2 结果与讨论
2.1 苏丹红Ⅳ的鉴别与确认
实验首先取10批龙血竭药材,按照2020年版《中国药典》四部色素指导原则进行薄层色谱鉴别,结果发现有2批样品在与苏丹红Ⅳ薄层色谱相同的位置上显相同颜色的斑点,但是斑点均在前沿,无法准确判断,然后将2批染色可疑的样品再次换展开剂进行展开,使其在较好的比移值范围内,并增加其它系列苏丹红染色剂从而更加准确地判断,结果发现苏丹红Ⅳ确实与1号和5号样品颜色一致。接着采用液相色谱法进行判断,供试品与苏丹红Ⅳ在相同的保留时间出现相同的色谱峰。然后通过紫外光谱图比较,液相色谱串联质谱进行确认和佐证,光谱图中供试品和苏丹红Ⅳ对照品均在518 nm处有最大吸收,且曲线的性状基本一致,质谱佐证实验中的m/z381.9 [M+H]为苏丹红Ⅳ的准分子离子峰,二级碎片离子分别为m/z223.8、208.0、142.4、90.7 四个碎片离子,且5号和10号两批样品的质谱二级碎片图与苏丹红Ⅳ标准品一致,因此更加确定此2批龙血竭药材进行了苏丹红Ⅳ的染色。
2.2 提取溶剂和色谱条件的选择
苏丹红Ⅳ易溶于笨、油脂和苯酚等极性较小的溶剂中,难溶于水,微溶与乙醇。目前主要有乙腈、正己烷和丙酮等溶剂进行提取的报道,食品中苏丹红的检测方法也是采用正己烷进行处理,以丙酮定容后进行测定,因此本实验对乙腈、正己烷和丙酮三种溶剂进行考察,发现乙腈提取色谱峰形较好,且乙腈比正己烷和丙酮毒性低,因此选择乙腈为提取溶剂。同时本实验参考文献报道和食品中苏丹红检测的国标方法,分别采用乙腈和水、乙腈和0.1%甲酸水溶液以及甲醇和0.1%甲酸水溶液作为流动相进行考察。结果发现乙腈和水溶液作为流动相,对照品的色谱峰有点拖尾;甲醇和0.1%水溶液作为流动相,苏丹红Ⅳ的出峰时间太长,峰形较宽;最后采用乙腈和0.1%甲酸水溶液时,色谱峰形对称,基线平稳,理论塔板数高。采用DAD检测器对苏丹红Ⅳ提取光谱图,结果发现其在518 nm下具有最大吸收,故本实验最终确定乙腈-0.1%甲酸水溶液为流动相,检测波长为518 nm,对苏丹红Ⅳ进行检测。
2.3 线性关系与检出限
取“1.3.1”项下的对照品储备溶液作为对照品5号溶液,采用逐级稀释法,制成含苏丹红Ⅳ分别 为 1.142、3.807、12.69、42.3、141 μg /mL 的 系列对照品工作溶液,分别作为1~5号对照品溶液,然后精密吸取上述1~5号对照品溶液各10 μL,按照1.2仪器工作条件进行测定,以苏丹红Ⅳ的质量浓度(μg/mL)为横坐标,相对应的色谱峰面积为纵坐标进行线性回归,计算得苏丹红Ⅳ的质量浓度在1.142~141.0 μg/mL范围内线性方程为y=53.237x-13.628,相关系数 r=0.999 9。
将1号对照品溶液进行稀释进样,当S/N=3时为检出限,得到苏丹红Ⅳ的检出限为2.28 ng/mL。
2.4 精密度试验
取同一批号200101的疑似染色的龙血竭药材,按照1.3.2法同时制备6份供试品溶液,然后按照1.2仪器工作条件进行测定。6份供试品中苏丹红Ⅳ含量测定值分别为 2.750 4、2.747 9、2.746 3、2.755 8、2.805 1、2.708 5 mg/g,平均值为 2.752 3 mg/g,计算得测定结果的相对标准偏差为1.12%,表明该方法具有良好的精密度。
2.5 稳定性试验
取同一批号200101疑似染色的龙血竭药材,按照“1.3.2”法制备供试品溶液,连续考察24 h,分别在 0、1、2、4、8、12、24 h 按照 1.2 仪器工作条件进行测定,记录色谱峰面积分别为1 147.2、1 146.1、1 127.1、1 156.3、1 168.7、1 165.2、1 182.9 mAU,平均值为1156.2 mAU,计算得测定结果的相对标准偏差为1.57%,表明该供试品溶液在24 h内稳定性良好。
2.6 回收试验
精密称取同一批号200101疑似染色的龙血竭药材0.1 g,共18份,每6份为一组,然后分别向上述3组样品中精密加入1.3.1配制的苏丹红Ⅳ对照品储备溶液 100、200、300 μL,按照 1.3.2制备供试品溶液,在1.2仪器工作条件下进行测定,结果见表2。样品加标色谱图见图6。由表2可知,样品加标回收率为94.72%~99.21%,说明该方法具有较高的准确度。

表2 加标回收试验结果(n=9)
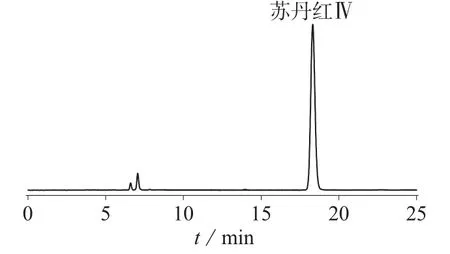
图6 样品加标回收色谱图
3 结语
首先按2020年版《中国药典》四部色素指导原则进行薄层色谱鉴别,结果发现10批不同产地的龙血竭药材中有2批药材疑似是进行了苏丹红IV的非法染色,且2批龙血竭药材薄层色谱均与其它批次药材斑点不一致,推测可能是其它树脂类药材直接进行染色冒充正品龙血竭药材。采用高效液相色谱串联质谱法对疑似染色的两批药材中苏丹红IV进行确认和测定,并进行方法学研究,稳定性、重复性和回收率均符合要求,并且确认两批药材进行了苏丹红IV染色。由于目前龙血竭药材的收录标准中未有非法染色的检查项目,且非法染色的苏丹红对人体伤害具有一定的致癌性,很大程度上威胁这患者的用药安全,难以保证药品的有效性,因此为了药材的的使用安全,建议将苏丹红的染色鉴别增加到龙血竭药材标准中。
