高效液相色谱-串联质谱法测定果汁及乳饮料中9种焦油色素
2021-12-02张琦李文君李墨浠牛悦齐胡婷婷芦春梅王准翟亚楠周晓
张琦,李文君,李墨浠,牛悦齐,胡婷婷,芦春梅,王准,翟亚楠,周晓
(长春海关,长春 130062)
焦油色素是以煤焦油为原料制成的,焦油色素制备成本低,颜色鲜艳,着色力强且稳定不易褪色,在食品加工业中被广泛采用。食用合成色素对人体可能具有致癌性,毒理学研究发现,某些合成色素有慢性毒性或致癌性,许多食用合成色素在生产过程中还可能混入有害重金属,人长期食用会产生毒害。因此各国都严格控制其使用范围和使用量[1-3]。
各国食用色素允许使用品种和范围的差异,使我国出口食品中的色素含量项目面临很大风险。美国儿童食品及药物检测机构公布了关于儿童食品禁止使用8种14个项目的食用焦油色素及食品添加剂标准的通知。韩国食品和药物管理局已签署了关于儿童食用色素(包括铝色淀)被禁止使用的文件。食用焦油色素的14个项目包括绿3、食用焦油色素、绿3铝湖、湖红40、湖红40铝色淀、蓝1、蓝2铝湖、青1、青1铝湖、青2、青2铝湖、黄4、黄4铝湖、黄5、黄5铝湖、红3以及红102等。我国允许使用新红、苋菜红、柠檬黄、靛蓝、亮蓝等食用合成色素,允许使用的食品种类有果味水、果味粉,糖果、糕点上的彩装、罐头等[4-7]。
目前国内外食品中焦油色素的检测方法主要有薄层色谱法、紫外分光光度法、导数分光光度法、示波极谱法、检测盒法、高效液相色谱法及高效液相色谱-质谱联用法[8-15]。薄层色谱法繁琐费时,测定结果偏低。紫外分光光度法、导数分光光度法这两种方法对于二组分体系较为有效,对于多组分混合体系,需要结合数学方法。示波极谱法已被用于检测饮料中柠檬黄、苋菜红、胭脂红三种焦油色素。检测盒法操作简单、快速,检测一个样品平均仅需要5~10 min,对几种常用焦油色素最低检出质量浓度均可达到2.0 mg/L,但高浓度的乙醇、淀粉以及蛋白质对色素的检测有一定的干扰作用。高效液相色谱法及高效液相色谱-质谱联用法可采用单一波长或双波长检测,结合梯度洗脱,能同时测定几种混合色素,方法准确,但必须结合样品预处理,否则不能有效排除干扰。笔者探讨并建立了固相萃取-高效液相色谱-串联质谱法同时测定果汁及乳饮料中酸性橙Ⅱ、酸性黄36、偶氮玉红、赤藓红、诱惑红、日落黄、喹啉黄、罗丹明B、柯衣定等9种焦油色素的分析方法。经过实验验证,该方法能够有效去除干扰物质,准确测定相应基质中目标焦油色素含量,可以为果汁及乳饮料中9种焦油色素的检测提供有效的技术支撑。
1 实验部分
1.1 主要仪器与试剂
高效液相色谱-串联质谱仪:API 4500型,美国AB公司。
超声振荡器:AS10200-20L型,天津奥特赛恩斯仪器有限公司。
分 析 天 平:(1)SQP型,感 量 为 0.1 mg;(2)AB265-S型,感量为0.01 g,梅特勒托利多仪器(上海)有限公司。
高速冷冻离心机:Allegra X30R型,德国贝克曼库尔特公司。
氮吹仪:MGS-2200型,日本东京理化公司。
PH计:PH3310型,德国WTW公司。
乙腈、甲醇:质谱级,美国Fisher公司。
甲酸、氨水:分析纯,上海国药集团化学试剂有限公司。
乙酸铵、铁氰化钾、柠檬酸:分析纯,天津市科密欧化学试剂有限公司。
酸性橙Ⅱ、酸性黄36、偶氮玉红、赤藓红、诱惑红、日落黄、喹啉黄、罗丹明B、柯衣定标准品:纯度不小于98%,德国Dr. Ehrenstorfer公司。
1.2 仪器工作条件
(1)液相色谱。色谱柱:Agilent Pursuit XRs 5 C18色谱柱(100 mm×2.0 mm,l3 μm,美国安捷伦科技有限公司);柱温:室温;进样体积:10 μL;流动相:A相为20 mmol/L的乙酸铵水溶液,B相为乙腈;流量:0.25 mL/min;流动相梯度洗脱程序见表1。

表1 流动相梯度洗脱程序
(2)质谱。电离方式:电喷雾(ESI);电喷雾电压:5 500 V/-4 500 V;检测方式:多反应监测(MRM);扫描方式:正/负离子扫描;气帘气(CUR)压力:0.31 MPa(45 psi);雾化气(GS1)压力:0.31 MPa(35 psi);辅助气(GS2)压力:0.31 MPa(35 psi);碰撞气(CAD)压力:0.15 MPa(6.0 psi);离子源温度:550℃;目标化合物的监测离子对、去簇电压和碰撞能量见表2。

表2 目标化合物的监测离子对、去簇电压和碰撞能量

续表2
1.4 样品处理
1.4.1 样品提取
称取样品2 g(精确到0.01 g)至50 mL离心管中,加入5 mL水混匀,加入沉淀剂(铁氰化钾)后,4 000 r/min离心3 min,转移上清液,用20%柠檬酸溶液调pH值至5,待净化。
1.4.2 样品净化
首先活化WAX固相萃取柱(6 mL/150 mg),依次加入6 mL甲醇、6 mL水活化固相萃取柱,将待净化液转移至固相萃取柱,依次用6 mL 0.5%甲酸甲醇溶液、6 mL水清洗,弃去全部流出液,加入5 mL 25%氨化甲醇洗脱至10 mL试管中,于40℃下氮吹至近干,用甲醇定容至2.0 mL,过0.22 μm滤膜后上机测定。
2 结果与讨论
2.1 样品净化方法的选择
根据焦油色素的分子结构特征,分别选取C18固相萃取柱、WAX固相萃取柱、HLB固相萃取柱进行样品加标回收试验,结果如表3。

表3 9种焦油色素不同固相萃取柱的回收率
HLB柱是反相固相萃取柱,吸附目标色素回收率为70%~80%,柯依定回收率低于60%;C18柱整体回收率为60%~80%;WAX柱具有弱阴离子交换及反相两种交换机理,9种目标色素整体回收率为80%~90%。由表3数据可知,WAX柱整体回收率均较高,9种目标物回收率稳定,因此选用WAX固相萃取柱。采用0.5%甲酸甲醇溶液淋洗固相萃取柱,可除去实际样品中部分天然色素。
2.2 液相色谱条件优化
分别以甲醇-水、甲醇-乙酸铵溶液、乙腈-水、乙腈-乙酸铵溶液作为流动相考察对目标物色素的洗脱效果。有机相用甲醇时色谱峰较宽,有机相用乙腈时色谱峰变窄,因此选择乙腈作为有机相。采用乙腈-水流动性体系,诱惑红、日落黄色谱峰拖尾现象严重,部分色谱峰分离度较差,水相添加20 mmol/L乙酸铵溶液后色谱峰变得尖锐,解决了峰间分离度不佳的情况,最终采用20 mmol/L乙酸铵溶液-乙腈作为流动相。
进行多目标物分析时,为满足分离度要求,需要目标物出峰时间适当延后,起始流动相采用80%乙酸铵溶液-20%乙腈保持3 min,利于较强极性杂质的洗脱;11 min内将乙腈比例由20%提高到80%,使多数目标物色素洗脱,20%乙酸铵溶液-80%乙腈保持2 min,将极性最弱的罗丹明B、柯衣定洗脱出来,最后平衡色谱柱回归起始流动相80%乙酸铵溶液-20%乙腈,保持4 min。
9种目标色素优化后的洗脱程序见表1,色素标准溶液色谱图见图1、图2。

图1 7种负扫描模式焦油色素总离子流图

图2 罗丹明B、柯衣定正扫描模式色谱图
2.3 质谱检测条件优化
首先用针泵将每种目标焦油色素单独进样,进行全扫描寻找母离子,优化去簇电压,确定母离子;再进行碎片离子扫描,寻找强度最大的几个特征离子,并优化碰撞能量,选择出合适的子离子,一般为2~3个,以所选母离子及其子离子进行MRM多反应监测扫描,并再次优化去簇电压和碰撞能量,使得所选离子对灵敏度最高。本研究选择了多对离子中强度高、灵敏度高的两对离子作为定性离子,为保证目标物的定量和定性的准确性,选择其中灵敏度最高的一对离子作为定量离子。测定时,可根据保留时间和两对离子对进行定性确证;采用标准物质和待测样品的MRM总离子流图中的色谱峰面积,用外标法定量。实验结果表明,本方法确定的特征目标检测离子测定灵敏度高,选择性好,干扰物少,线性范围宽,重现性好,定量准确,阳性确证结果准确可靠。
2.4 线性方程、线性范围与检出限
在本研究所确定的LC-MS/MS检测条件下,分别对多种焦油色素标准物质在不同浓度范围内制备5个不同浓度水平的标准溶液进行测定,以目标色素的质量浓度为横坐标(x)、以定量离子色谱峰面积为纵坐标(y)绘制标准工作曲线,计算目标色素线性回归方程及相关系数r。根据各焦油色素定性离子信噪比S/N≥3计算方法检出限。方法的线性范围和检出限见表3。

表3 线性方程、线性范围和检出限
2.5 精密度和回收试验
选择未检出酸性橙Ⅱ、酸性黄36、偶氮玉红、赤藓红、诱惑红、日落黄、喹啉黄、罗丹明B、柯衣定等9种焦油色素的样品,分别添加 0.5、1.0、5.0 μg/kg的标准品进行回收试验,每个浓度做6次平行试验,计算方法的平均回收率和精密度,结果见表4。样品加标色谱图分别见图3、图4。

图3 7种负扫描模式焦油色素加标色谱图
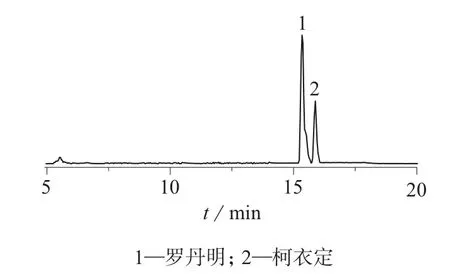
图4 2种正扫描模式焦油色素加标色谱图

表4 精密度和回收试验结果
由表4可知,焦油色素的回收率为78.00%~97.08%,相对标准偏差为4.49%~11.27%。回收率和相对偏差均在允许范围内。
3 结语
建立了固相萃取柱-高效液相色谱-串联质谱法测定果汁和乳饮料中酸性橙Ⅱ、酸性黄36、偶氮玉红、赤藓红、诱惑红、日落黄、喹啉黄、罗丹明B、柯衣定等9种焦油色素的分析方法。本方法利用固相萃取柱对样品中的焦油色素进行富集,有效去除基体干扰,以选择离子对待测物定性,用外标法进行定量,开发出了一个高效、快速、准确、可同时检测多种焦油色素的分离和分析确证方法。该方法检测下限完全满足国内外最高残留限量对食品的检测要求,能够为控制非法添加焦油色素的食品流入销售市场提供有效的保障。随着食品工业的发展,更多新型焦油色素被非法用于食品中,继续完善焦油色素准确、快速检测方法,对于保障人民身体健康、提高进出口监管的针对性和有效性,保障进口食品安全,将起到重大意义。
