H2O分子在Cu(110)和Ni(110)表面吸附与解离行为的理论研究
2021-12-02齐昶霖侯雪捷刘绍丽
齐昶霖,侯雪捷,刘绍丽
(烟台大学 化学化工学院,山东 烟台 264005)
1 研究背景与意义
大多数暴露在大气条件下的金属表面都覆盖着一层薄薄的水,水还涉及界面的许多物理和化学过程,包括电化学和光化学过程、多相催化中的表面反应、金属腐蚀和环境科学等[1-3].其中,金属腐蚀是金属和周围环境发生化学或电化学反应而导致的一种破坏性侵蚀,具有极大的破坏性,直接影响金属构件的使用寿命,甚至造成严重的事故.因此,研究金属腐蚀在分子层面上的反应机理,即水和金属表面的相互作用,确定水分子和金属反应的过程尤为重要.
在过去的报道中,人们已经使用传统的表面科学技术对低温下水的吸附和反应进行了大量研究.其中很多研究集中在Cu和Ni的低指数面上.之前的研究发现,在Ni(110)表面上,水以分子态形式吸附[4-6].Jacobi等[7]发现,H2O至少在前两层显示出层增长,且得出单层最大覆盖率为 0.7 ML.Pangher等[8]研究表明,水分子在Ni表面吸附,通过氧端与表面键合,O—Ni键的键长为(2.06±0.03)Å,且H—O—H平面相对于表面法线倾斜.实验结果表明,水分子在c(2×2)结构中不形成团簇[8-11].最低温度为 205 K 时,观察到水发生部分解离[10].Benndort等[12]研究了基底几何形状及预先吸附的氧气对Ni(110)表面H2O结构和反应性的影响.他们发现,在 80 K 时水以分子态吸附,而当T>200 K 时水会部分解离.热脱附显示了4个峰,分别是冰层形式的水脱附(T=155 K),多分子水簇的脱附(T=210 K),被OH稳定的水二聚体的脱附(T=245 K),以及由于OH歧化而引起的水脱附(T=360 K).预先覆盖的氧气有利于水的吸附和解离[4-6,12].Nakamura等[13]观察到在Ni(110)表面,水单体或团簇分子在低至 150 K 的温度下解离.在Ni(111)表面上,水单体(D2O)在低覆盖率下位于Ni原子的顶部,而随着覆盖率的增加(0.03<θ<0.33),环状六聚体的水团簇分子(D2O)6在表面生长.在T>165 K 时,吸附的水分子分解产生OD基团.
与Ni表面相同,许多实验研究表明,在低温下水以分子态形式吸附在Cu表面[14-17].而在Cu表面吸附的水分子是否解离存在巨大争议.Spitzer等[18]研究表明,在Cu(110)表面,90 K 时水作为分子吸附,90~300 K 时H2O解离为O原子和OH基团.Polak等[19]也发现在190 K左右,Cu(110)表面吸附的水分子部分解离.但Bange等[20]和Lackey等[21]在 100 K 时没有观察到吸附水分子的解离.Ren等[22-23]研究了Cu(110)面上的水覆盖层,结果发现单个水分子在顶位稳定吸附,Cu—O键长为2.18Å.覆盖度为 1 ML 时水分子可以形成冰双层结构,H-down双分子层比H-up双分子层更稳定.他们还研究了Cu(110)表面上的半解离水覆盖层,与未解离的水覆盖层相比,吸附能增加,但功函也增加,说明水分子在Cu(110)表面更倾向于分子态吸附.在Cu(111)和Cu(100)面上,水单体通过氧端结合到Cu原子的顶部,且水分子平行于金属表面,吸附较弱[24].Tang等[24]的DFT计算表明,水单体在Cu(111)和Cu(100)面上的吸附能(~20 kJ/mol)约为实验值的一半(~40 kJ/mol)[14,25],这表明即使在低温和低覆盖度下,水分子也可能形成团簇.
尽管对于水和金属表面相互作用已经进行了大量的理论和实验研究,但对于水分子在不同表面上的解离机理并没有详细阐明,因此研究了水分子在Cu(110)和Ni(110)表面上的吸附与解离行为,确定了表面物种的稳定吸附构型,进一步比较了水的解离机理和吸附物种与表面的电荷转移(CT).该研究结果为进一步理解和研究水与金属表面之间的相互作用提供了有利的理论基础.
2 计算方法与模型
所有的计算都是在从头模拟程序包VASP中使用平面波赝势方法完成的[26-27].使用自旋极化的广义梯度近似和Perdew-Burke-Ernzerhof泛函(GGA-PBE)描述了交换能和相关能[28].过渡态的计算使用VASP软件中的CI-NEB线性插值方法[29].当作用在原子上的力小于 0.01 eV/Å 时,完成几何优化.对于每个优化的固定点,均在相同的理论水平上进行振动分析,以确定其性质(能量极小点或鞍点).使用面心立方(fcc)晶胞优化晶格参数,并使用Monkhorst-Pack方法自动生成的15×15×15 k 点网格对它的倒易空间进行采样[30].
对于Cu(110)和Ni(110)表面,使用真空区域宽度为10.0Å的周期性平板,用3×3×1 Monkhorst-Pack k点采样和 380 eV 的截断能进行表面结构弛豫和总能量计算.使用(2×2)表面尺寸对周期表面进行建模.使用了4层模型,吸附物种及上面2层弛豫,而底部两层固定.如图1所示,Cu(110)面和Ni(110)面上可能的吸附位点为顶位(top,T),桥位(bridge,B),长桥位(long bridge,LB)和短桥位(short bridge,SB).
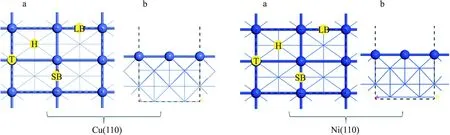
图1 Cu(110)面和Ni(110)面的俯视图(a),侧视图(b)及可能的吸附位
使用公式1计算吸附能,其中EX/slab是吸附体系的总能量,Eslab是清洁金属表面的总能量,EX是气相中游离吸附物的能量.因此,Eads越负,吸附越强.由于零点能对表面反应影响很小并且主要影响气体分子,因此所报道的吸附能没有考虑零点能的校正.
Eads=EX/slab-Eslab-EX.
(1)
根据公式2和3计算解离势垒(Ea)和反应能(Er),其中EIS,ETS和EFS是相应的初始状态(IS),过渡状态(TS)和最终状态(FS)的能量.
Ea=ETS-EIS.
(2)
Er=EFS-EIS.
(3)
3 结果与讨论
3.1 表面物种H,O,OH和H2O的吸附
在Cu(110)和Ni(110)表面,首先考察表面物种H、O、OH、H2O在表面的稳定吸附位及吸附构型.表1列出了表面物种在不同吸附位点的吸附能,稳定吸附构型如图2所示.
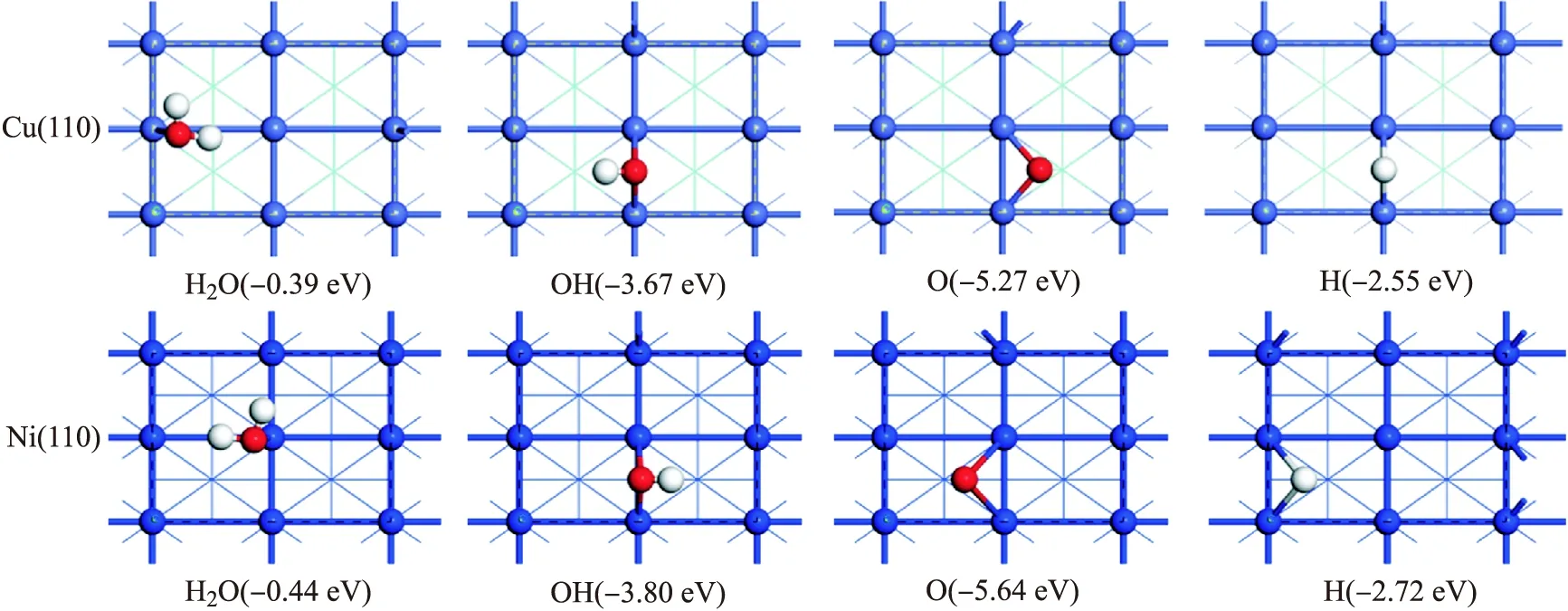
图2 H2O,OH,O和H在Cu(110)面和Ni(110)面的稳定吸附构型及吸附能

表1 表面物种在Cu(110)面和Ni(110)面上不同吸附位点的吸附能 eV
在Cu(110)表面,H原子在短桥位稳定吸附,吸附能为 -2.55 eV,Cu—H距离为1.651和1.651Å.如表2所示,洞位和长桥位的H原子吸附能分别比短桥位少0.06和 0.17 eV.H原子在顶位吸附的吸附能为 -1.99 eV,远小于短桥位,吸附稳定性相对较差.对于O原子,最稳定的吸附位为洞位,吸附能为 -5.27 eV,Cu—O距离为1.882、1.882、2.940和 2.940 Å.长桥位,短桥位和顶位的O原子吸附能分别比洞位少0.16,0.38和 1.63 eV.优化后的OH物种在短桥位稳定吸附,吸附能为 -3.67 eV,Cu—O距离为2.036和 2.036 Å,与表面的夹角为39.34°.OH物种在长桥位和顶位的吸附能比短桥位少0.34和 0.71 eV,吸附稳定性相对较差.洞位的OH物种优化后迁移到短桥位.最后,对于H2O分子的吸附,研究了所有高对称吸附位点.优化后,H2O在顶位稳定吸附,吸附能为 -0.39 eV,Cu—O距离为 2.178 Å,与Cu(110)面平行.H2O分子在短桥位的吸附能为 -0.27 eV,吸附不稳定.这一结果与文献[23-24]报道一致.
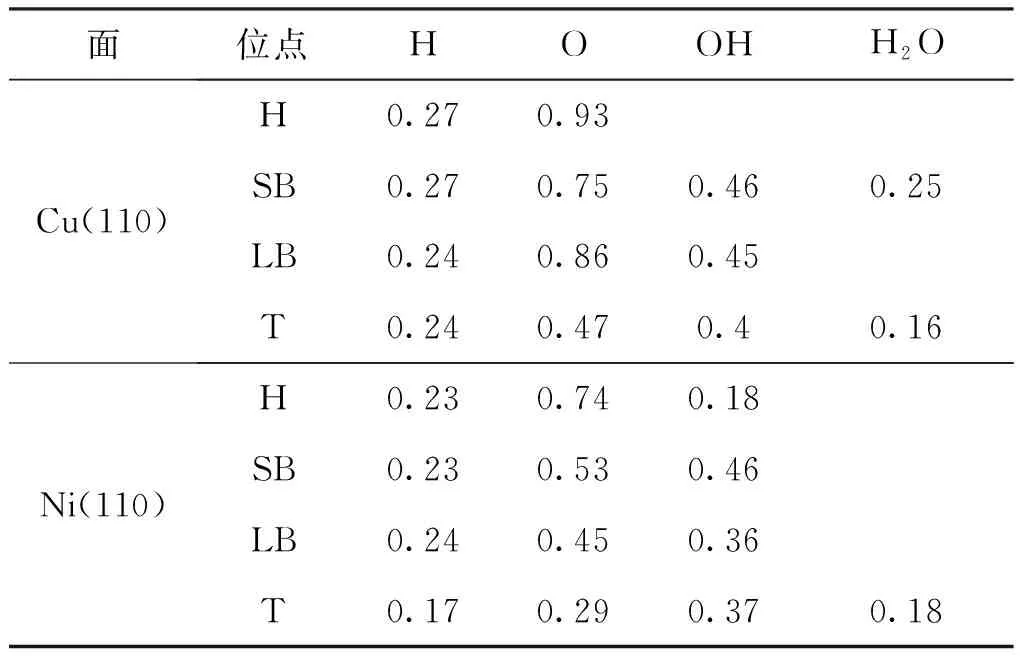
表2 Cu(110)和Ni(110)表面上的表面物种吸附前后的电荷转移(CT/e)
在Ni(110)表面,H原子在接近洞位的位点稳定吸附,偏转洞位上方,靠近短桥位,吸附能为 -2.72 eV,Ni—H键距离为1.699、1.702、1.803和1.805Å.短桥位和长桥位的H原子吸附能分别比洞位少0.05和 0.07 eV.Seenivasan等[31]报道的H原子在短桥位稳定吸附,吸附能为 -2.64 eV,与长桥位的吸附能(-2.63 eV)相似.对于O原子,洞位(-5.64 eV)的吸附能大于短桥位(-5.55 eV),吸附更稳定.O原子在长桥位的吸附能为 -5.33 eV,该构型是O原子从一个洞位转移到另一个洞位的过渡态,转移的能垒为 0.31 eV.O原子在顶位的吸附能为 -4.32 eV,吸附稳定性相对较差.OH物种在短桥位稳定吸附,吸附能为 -3.80 eV,Ni—O距离为1.917和 1.919 Å,与表面的夹角为23.98°.OH物种在长桥位的吸附能为 -3.18 eV,与顶位的吸附能(-3.12 eV)相似,Ni—O距离为2.076和 2.077 Å,吸附稳定性相对较差.洞位的OH物种吸附能仅为 -2.88 eV,Ni—O距离为2.320、2.340、2.358、2.378 Å.与Cu(110)面类似,不同位点不同构型的H2O分子优化后在顶位稳定吸附,且平行于Ni(110)表面,吸附能为-0.44 eV,Ni—O键距离为 2.125 Å.这一结果与Seenivasan等[31]的报道一致.
上述结果表明,H2O分子以平行于表面的构型吸附在Cu和Ni表面原子的顶位.在紧密堆积的Cu、Ni的(111)面及(100)面上,H原子,O原子以及OH物种倾向于吸附在深洞位和洞位上[32-36].但在(110)面,由于金属原子之间距离增加,OH物种更倾向于在短桥位吸附.表面物种在Ni(110)面的吸附能大于Cu(110)面,表明表面物种在Ni表面吸附更稳定.
表2中Bader电荷分析获得的在吸附物和基底之间的电荷转移的值表明,基底充当电子给体,而吸附物充当电子受体.在Cu和Ni的(110)表面上,电荷转移按以下顺序减少:O> OH> H,这与Cu和Ni的金属特性和被吸附物的电负性有关.对于不同吸附位点的同种吸附物,电荷转移的量与吸附稳定性的顺序密切相关.对于OH和O,电荷转移越多,吸附越稳定.
3.2 H2O分子的解离路径
基于表面物种的最稳定吸附构型,我们计算了Cu(110)和Ni(110)表面上的H2O分子解离为OH,O和H的反应路径.初始状态(IS),过渡状态(TS)和最终状态(FS)的几何构型如图3所示.TS的反应能垒(Ea),反应热(Er)和结构参数如表3所示.

图3 Cu(110)面和Ni(110)面上H2O解离反应稳定中间体的构型

表3 Cu(110)和Ni(110)表面上H2O解离反应的反应能垒(Ea, eV),反应热(Er, eV)和断裂的O—H键距离(dO-H,Å)

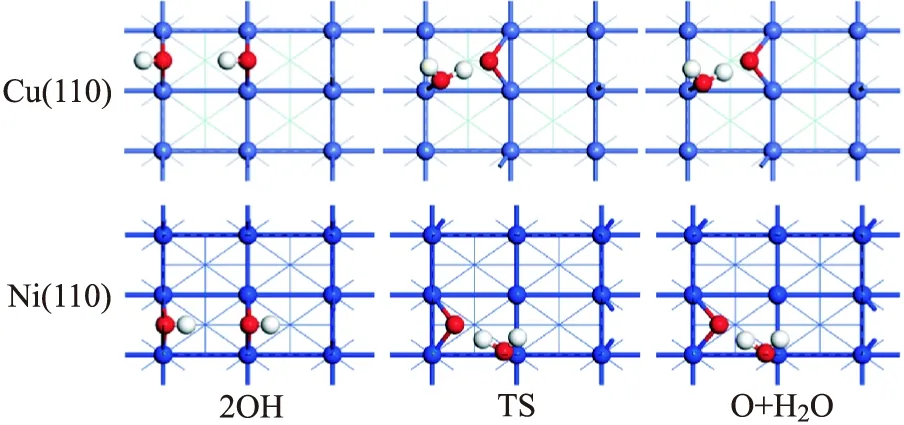
图4 Cu(110)面和Ni(110)面上OH歧化反应稳定中间体的构型
4 结语
文中利用密度泛函理论方法研究了在Cu(110)和Ni(110)表面上H,O,OH和H2O的吸附以及H2O分子的解离反应.
在Cu(110)面上,H原子和OH物种在短桥位稳定吸附,O原子在洞位稳定吸附.在Ni(110)表面上,H原子和O原子在洞位稳定吸附,OH物种在短桥位稳定吸附.H2O分子以平行于表面的构型吸附在Cu和Ni的顶位.对于不同吸附位点的同种吸附物,电荷转移的量与吸附稳定性有关.在Cu和Ni的(110)表面上,电荷转移按以下顺序减少:O> OH> H,这与Cu和Ni的金属特性和被吸附物的电负性有关.
对于H2O的解离反应,H2O分子可以解离为OH+H,而OH继续解离需要高的反应能垒,反应难以发生.OH歧化反应的反应能垒远大于其逆反应,因此在Cu(110)和Ni(110)表面上,水分子的最终解离产物为OH和H.这些结果可以为进一步了解和研究水与金属表面之间的相互作用提供有利的理论基础.
