利用异源表达挖掘纤维堆囊菌So0157-2的新型天然产物
2021-11-29周海波申琪瑶陈汉娜王宗杰李越中张友明卞小莹
周海波,申琪瑶,陈汉娜,王宗杰,李越中,张友明,卞小莹
(山东大学,微生物技术国家重点实验室,山东 青岛 266237)
黏细菌属于革兰氏阴性细菌,是新颖活性天然产物的重要来源,具有巨大的药用开发潜力。目前已从黏细菌中发现了大约100多种次级代谢产物的基本结构和600多种结构类似物。这些化合物不仅结构类型丰富,包括聚酮类、非核糖体肽、萜类以及其他杂合的结构类型,同时也显示了广泛的生物活性,如抗菌、抗肿瘤、抗病毒、免疫抑制、抗疟等[1-6]。
随着基因组测序技术的发展,越来越多的基因组信息分析表明黏细菌基因组所蕴藏的生物合成基因簇(biosynthetic gene cluster,BGC)合成新颖次级代谢产物的潜力远远超出了目前已分离获得的化合物的数目。例如,抗肿瘤药物埃博霉素(epothilone)的产生菌纤维堆囊菌So0157-2(Sorangium cellulosumSo0157-2)的基因组测序结果表明,该菌株拥有14.78 Mb 的环状染色体,是目前已知基因组最大的原核生物[7-9]。除了已知的埃博霉素系列衍生物[10-15]之外仍未见其他化合物从该菌株中分离报道,这预示着该菌株仍具有较大的代谢潜能。对隐性基因簇进行有效的激活和改造,能够为药物先导结构的发现提供更多的化合物资源。由于纤维堆囊菌So0157-2 生长相对较慢、培养困难,本源菌也缺乏合适的遗传操作体系,对其天然产物的开发存在着诸多挑战。
近年来天然产物生物合成基因簇的异源表达策略受到了越来越广泛的关注。异源表达是将完整的基因簇克隆到穿梭载体上然后转移到合适的异源宿主中进行表达,是挖掘难培养或未培养微生物次级代谢产物的有效手段。相比于原始宿主,异源宿主在遗传操作和生长速度上有较大优势。一方面,异源表达可以用于提高次级代谢产物的产量。另一方面,异源表达可以用于鉴定已知天然产物的生物合成途径。此外,异源表达还可以用于鉴定隐性基因簇,发现新型天然产物,特别有利于难培养或未培养微生物次级代谢产物的挖掘[16-19]。
异源表达的难点之一在于大部分天然产物生物合成基因簇都相当大(>10 kb),PCR 扩增很难得到如此长的DNA 序列。通过构建和筛选基因文库虽然能够获得目的基因簇,但是工作量非常大,且不一定能获得完整的基因簇。随着基因编辑技术的发展和进步,可以利用直接克隆技术(direct cloning)从基因组中克隆大型基因簇,比如LLHR(linear-linear homologous recombination)、 TAR(transformation-associated recombination)、CATCH(Cas9 assisted targeting of chromosome segments)等[20]。其中LLHR是由本团队于2012年开发的基于Red/ET 重组工程技术(recombineering)的直接克隆技术[21],并与异源表达相结合,广泛应用于天然产物基因簇的挖掘。2018年将Red/ET重组工程技术与核酸外切酶体外处理技术相结合,开发了ExoCET(exonuclease combined with RecET recombination)克隆技术,进一步提高了BGCs 直接克隆的效率[22]。此外,Red/ET 重组工程技术还可以用于基因簇的无痕定点突变,结构域替换等等,极大促进了大型基因簇的遗传操作[22-28]。
异源表达的另一个难点在于异源宿主的选择。大肠杆菌和酿酒酵母都是表征良好、易于遗传操作的模式菌株,为细菌和真菌中天然产物的表达提供了良好的异源宿主[29-31]。研究者也发现,当宿主菌与包含目标基因簇的菌株在进化上相近时,异源表达就更易成功。所以,很多链霉菌被开发为异源宿主菌,用于放线菌中基因簇的异源表达[32-33]。可用于黏细菌异源表达的异源宿主菌相对缺乏,目前常用的有黄色黏球菌(Myxococcus xanthus)、恶臭假单胞菌(Pseudomonas putida)以及Schlegelella brevitaleaDSM 7029(曾用名:BurkholderialesStrain DSM 7029,[Polyangium]brachysporumDSM 7029)等[16-18]。除了黄色黏球菌和S.brevitaleaDSM 7029之外,其他异源宿主产量普遍较低。黄色黏球菌也属于黏细菌属,自身生长周期慢、难于进行遗传操作等劣势也限制了其应用。S.brevitaleaDSM 7029 是一株能够产生多种非核糖体肽、聚酮-非核糖体肽杂合化合物的革兰氏阴性细菌,相较于黄色黏球菌具有生长快、易操作、代谢背景清晰等诸多优点,特别是在表达纤维堆囊菌来源埃博霉素中显示了作为通用底盘菌的潜力[34-37]。因此,该菌株可作为本研究的异源宿主候选菌。
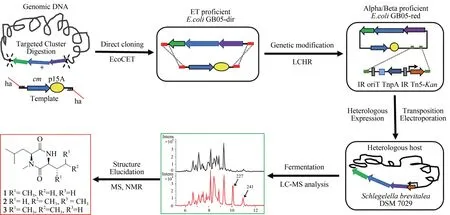
基于纤维堆囊菌So0157-2 基因组信息预测分析所显示出的代谢潜能,本论文利用ExoCET 直接克隆技术从该菌基因组DNA 中克隆了1 个未知功能的NRPS-PKS 杂合的基因簇BGC18。通过启动子插入,并以S.brevitaleaDSM 7029为异源宿主进行异源表达,经过液质联用(HPLC-MS)分析,正相与反相色谱柱靶向分离纯化,获得了3个该基因簇对应的表达产物。最后,通过核磁共振(NMR)结构鉴定和Marfey 反应确定了3 个化合物分别为新天然产物Cyclo(N-Me-L-Leu-L-Val)(1)和 Cyclo (N-Me-L-Leu-L-Leu)(2), 新 化 合 物Cyclo(N-Me-L-Leu-L-Ⅰle)(3)。将化合物结构与生物合成基因簇对比分析发现化合物结构的多样性来源于第1 个腺苷化结构域(A domain)对底物识别的宽泛性。此外,推测由于缺少硫醇化结构域(T domain)导致PKS 模块被跳过,从而只获得了两个NRPS模块指导合成的环二肽产物。该研究成功构建了基于Red/ET 重组工程技术的纤维堆囊菌So0157-2 基因簇的直接克隆、遗传修饰和异源表达体系,不仅丰富了纤维堆囊菌So0157-2 的代谢产物库,也为挖掘该菌株中其他新颖的天然产物用于药物筛选评价奠定了基础。
1 材料和方法
1.1 材料
1.1.1 菌株、质粒和引物
本研究所用的菌株和质粒见表1。寡核苷酸引物均由生工生物工程(上海)股份有限公司合成,本研究所用的寡核苷酸序列见表2。

表1 本研究所用的菌株和质粒Tab.1 Strains and plasmids used in this study

表2 本研究所用的引物Tab.2 Primers used in this study
1.1.2 培养基
LB 培养基:酵母粉0.5%,蛋白胨1%,NaCl0.1%,pH 7.0。
CYMG 培养基:胰蛋白胨0.8%,酵母粉0.4%,MgCl2·2H2O 0.4%,甘油5 mL/L。
M26 培养基:土豆淀粉0.8%,葡萄糖0.2%,蛋白胨0.2%,酵母提取物0.2%,CaCl2·2H2O 0.1%,微量元素液1 mL/L;pH 7.0。
VY/2 培 养 基 : 鲜 酵 母 0.5%, MgSO4·7H2O 0.4%,维生素B120.005%,CaCl2·2H2O 0.1%。
以上固体培养基均添加1.5%的琼脂。
1.1.3 主要试剂和仪器
限制性DNA 内切酶、T4 DNA 聚合酶购自New England BioLabs 公司;PrimeSTAR Max DNA聚合酶、DNA Marker 购自Takara 公司;卡那霉素(kanamycin,km)、氯霉素(chloramphenicol,cm)购自上海生工生物工程有限公司;培养基组分购自北京索莱宝生物科技有限公司;分析纯甲醇、无水乙醇、异丙醇购自国药集团化学试剂有限公司;色谱级甲醇、乙腈购自Thermo Fisher 科技有限公司。
液质联用仪型号为Thermo Fisher UltiMate3000与Bruker Amazon SL联用;高效液相色谱仪型号为Agilent 1260;电转仪型号为Eppendorf AG 4309;核磁共振波普仪型号为Agilent 500 MHz DD2;HPLC 制备所用色谱柱型号为Agilent ZORBAX SB-C18,9.4 mm×250 mm,5 μm;液质分析所用色谱柱为Thermo Scientific Acclaim RSLC 120 C18,2.1 mm×100 mm,2.2 μm。
1.2 BGC18 基因簇的直接克隆与异源表达
1.2.1S.cellulosumSo0157-2基因组提取
将-80 ℃保藏的S. cellulosumSo0157-2 接种到VY/2 平板上,30 ℃恒温培养,待长出菌膜,转接到表面湿润的M26平板上。刮取M26平板培养5~7 d的菌落置于50 mL离心管中,水洗两次,然后根据菌量加入适量的无菌水,涡旋混匀。吸取1.8 mL菌液分装到2 mL EP 管中,12 000 r/min 离心1 min,弃上清。加入450 μL Tris-HCl(10 mmol/L,pH 8.0),吹打混匀。加入30 μL 20 mg/mL 蛋白酶K,颠倒混匀。加入40 μL 10%SDS(sodium laurylsulfonate)后,轻轻混匀。50 ℃水浴1~2 h,中间间断颠倒直至溶液变澄清。加入500 μL 苯酚-氯仿-异戊醇(25∶24∶1),快速混匀,至溶液呈乳浊状,13 800 r/min 离心15 min。用去尖的移液吸头吸取300 μL 上清置于新的 2 mL EP 管中。加入 35 μL 3 mol/L NaAc(pH 7.5),混匀后,加入1.2 mL 无水乙醇,混匀。准备新的2 mL EP 管并加入1 mL 70%乙醇,用黄色吸头将悬浮的DNA 挑至该EP管中。10 000 r/min 离心1 min。弃上清,倒置于吸水纸上并用吸水纸将管壁上的水吸掉。室温干燥15~20 min。加入200 μL 双蒸水(double-distilled H2O,ddH2O),放置4°C冰箱备用。
1.2.2S.cellulosumSo0157-2基因组酶切产物制备
将基因组DNA 利用限制性内切酶DraⅠ和HindⅢ酶切,释放目标基因簇片段。取上述制备的基因组 DNA 200 μL,加入 40 μL 10×Cutsmart buffer、 12 μL 限 制 性 内 切 酶DraⅠ 和Hind Ⅲ 、1.5 μL RNase A,用ddH2O 补齐至 400 μL,37 ℃反应3 h。取10 μL 酶切产物跑琼脂糖凝胶电泳进行检测。检测之后用等体积的苯酚-氯仿-异戊醇(25∶24∶1)抽提酶切产物除去蛋白,然后进行乙醇沉淀。
1.2.3 直接克隆载体的制备
以 BGC18-HAF 和 BGC18-HAR 为 PCR 引 物 ,以质粒p15A-cm-tetR-tetO-hyg-ccdB 为PCR 模板,进行PCR 扩增。取2 μL PCR 产物进行电泳检测,确认正确后将剩余PCR产物切胶回收目的片段。
1.2.4 目的基因簇BGC18的直接克隆
直接克隆载体和酶切基因组产物在体外用T4 DNA Polymerase 退火:取 200 ng 克隆载体、2 μL 10×NEB Buffer 2.1、0.13 μL T4 DNA Polymerase 混匀后,轻轻加入12 μL 基因组酶切产物,再用ddH2O将酶切体系补足到20 μL。PCR反应程序为:25 ℃,60 min;75 ℃,20 min;50 ℃,30 min;4 ℃保温。反应完成之后将产物室温除盐40 min。将上述除盐后的产物电转化到经L-Ara 诱导的感受态细胞E.coliGB05-dir/pSC101-BAD-ETgA 中。从LB 转化平板上挑取单菌落,小量提取质粒,进行酶切分析。
1.2.5 目的基因簇BGC18的遗传修饰
分别对质粒p15A-cm-BGC18 进行转座元件和启动子的插入。利用带有同源臂的引物对ⅠR-Pkm-C18-ⅠR-HAF1/HAR1对质粒pR6K-oriT-TnpA-ⅠR-km进行扩增,得到oriT-ⅠR-PkmPCR产物,PCR产物通过切胶回收纯化;随后将回收的产物与质粒p15A-cm-BGC18 共同转入E. coliGB05-red 感受态细胞中,使用合适的抗生素(km/cm)筛选重组子;重组质粒通过限制性内切酶ApaL Ⅰ进行酶切鉴定及测序鉴定。
1.2.6 目的基因簇BGC18异源表达
S.brevitaleaDSM 7029已作为异源宿主表达了多个黏细菌来源的基因簇,所以本研究中选择野生型的S.brevitaleaDSM 7029 作为首选异源宿主。首 先 将 质 粒 p15A-oriT-TnpA-ⅠR-Pkm-BGC18 ( 约1 μg)电转入野生型S.brevitaleaDSM 7029 中[24],通过含卡那霉素的CYMG 平板筛选重组子。分别利用detect-C18 in 7029-F1/R1,detect-C18 in 7029-F2/R2,detect-C18 in 7029-F3/R3,detect-C18 in 7029-F4/R4,detect-C18 in 7029-F5/R5五对引物对重组子进行菌落PCR鉴定。
1.2.7 菌株的发酵提取分离与LC-MS检测
将正确的重组子接种于含有50 mL CYMG 培养基(km 3μg/mL)的300 mL 锥形瓶中,30 °C,180 r/min 培养过夜制备种子液。转接50 μL 种子液于相同的培养基相同培养条件下培养2 d,然后每瓶加入1 mL经前处理的XAD16大孔树脂,继续恒温摇床培养2 d。
将菌体和大孔树脂通过100目筛进行分离,并用双蒸水将树脂洗涤3 次(尽量去除菌体)。将树脂倒入新的干燥的锥形瓶中,加入50 mL 的甲醇,30°C,180 r/min浸泡1 h。通过滤纸过滤将树脂和甲醇分离,并将甲醇组分减压浓缩蒸干得到粗提物。加入1 mL 色谱甲醇或乙腈溶解粗提物,然后12 000 r/min,离心 10 min,取上清过 0.22 μm 微孔滤膜并转移至HPLC 进样管中,待HPLC-MS 进行质谱检测。
液质检测条件如下:液质分析色谱柱;流动相,A相为水+0.1%甲酸,B相为乙腈+0.1%甲酸;流速0.3 mL/min;进样 3 μL;检测波长 190~400 nm;洗脱程序0~3 min,5% B;3~18 min,5%~95%B;18~22 min,95% B;22~25 min,5% B。质谱检测条件:电喷雾离子源,正离子模式,二级质谱AutoMS2,检测范围m/z70~2200。
1.2.8 氨基酸绝对构型的确定
采用Marfey 法将化合物1~3 分别进行酸水解, 水解产物分别与 L-FDAA (1-fluoro-2-4-dinitrophenyl-5-L-alanine amide) 或 D-FDAA 反应[35]。采用同样的方法制备标准品N-Me-L-Leu 与L/D-FDAA 的衍生产物,L/D-Val、L/D-Leu、L/D-Ⅰle、L/D-allo-Ⅰle 与 L-FDAA 的衍生物,随后进行LC-MS 分析。N-Me-L-Leu、L/D-Val 与 L/D-FDAA的衍生产物的分析条件与1.2.7液质检测条件一致,检测波长 330 nm。L/D-Leu、L/D-Ⅰle、L/D-allo-Ⅰle的衍生产物洗脱程序为0~45 min,5%~50% B。对应衍生物的参照分子量为[M+H]+m/z398(N-Me-L-Leu)、370(L/D-Val)、384(L/D-Leu,L/D-Ⅰle,L/D-allo-Ⅰle)。
2 结果和分析
2.1 S.cellulosum So0157-2生物信息学分析
通过antiSMASH 分析,S.cellulosumSo0157-2基因组(NCBⅠ数据库登录号:CP003969.1)共包含35 个BGCs(表3),编码聚酮(PKS)、非核糖体 肽 (NRPS)、 PKS-NRPS 杂 合 以 及 萜 类(terpene)等多种结构类型化合物。除了已知的epothilone[8]及另外两个萜类生物合成基因簇geosmin[40]和 eremophilene[41]之外,其余基因簇与目前已知的生物合成基因簇都存在着较大的不同,相似度较低,这预示着该菌株仍具有较大的代谢潜能。PKS-NRPS杂合的化合物通常具有新颖的结构以及广泛的生物活性[42],因此优先选取该类型基因簇进行激活。BGC18 预测为PKS-NRPS杂合基因簇,长度约为26.7 kb(图1)。核心基因(SCE1572_24725)包含3 个模块共有10 个结构域(C1-A1-T1,C2-A2-MT-T2,KS-AT-TE)(图 1)。其中A1(adenylation)被预测可能识别缬氨酸/丙氨酸/甘氨酸/亮氨酸/异亮氨酸(Val/Ala/Gly/Leu/Ⅰle),A2被预测可能识别亮氨酸,然后被N-甲基化形成N-甲基亮氨酸(N-Me-Leu),AT(acyltransferase)则可能识别丙二酰辅酶A(Malonyl-CoA),但是缺少PKS 所必需的硫醇化(thiolation,T)结构域,这些底物能否通过NRPS和PKS的延伸单元被依次加载、缩合,形成的终产物最后被硫酯酶(TE)结构域释放是一个值得研究的问题。
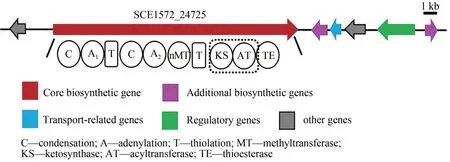
图1 BGC18基因簇结构Fig.1 Organization of the biosynthetic gene cluster BGC18
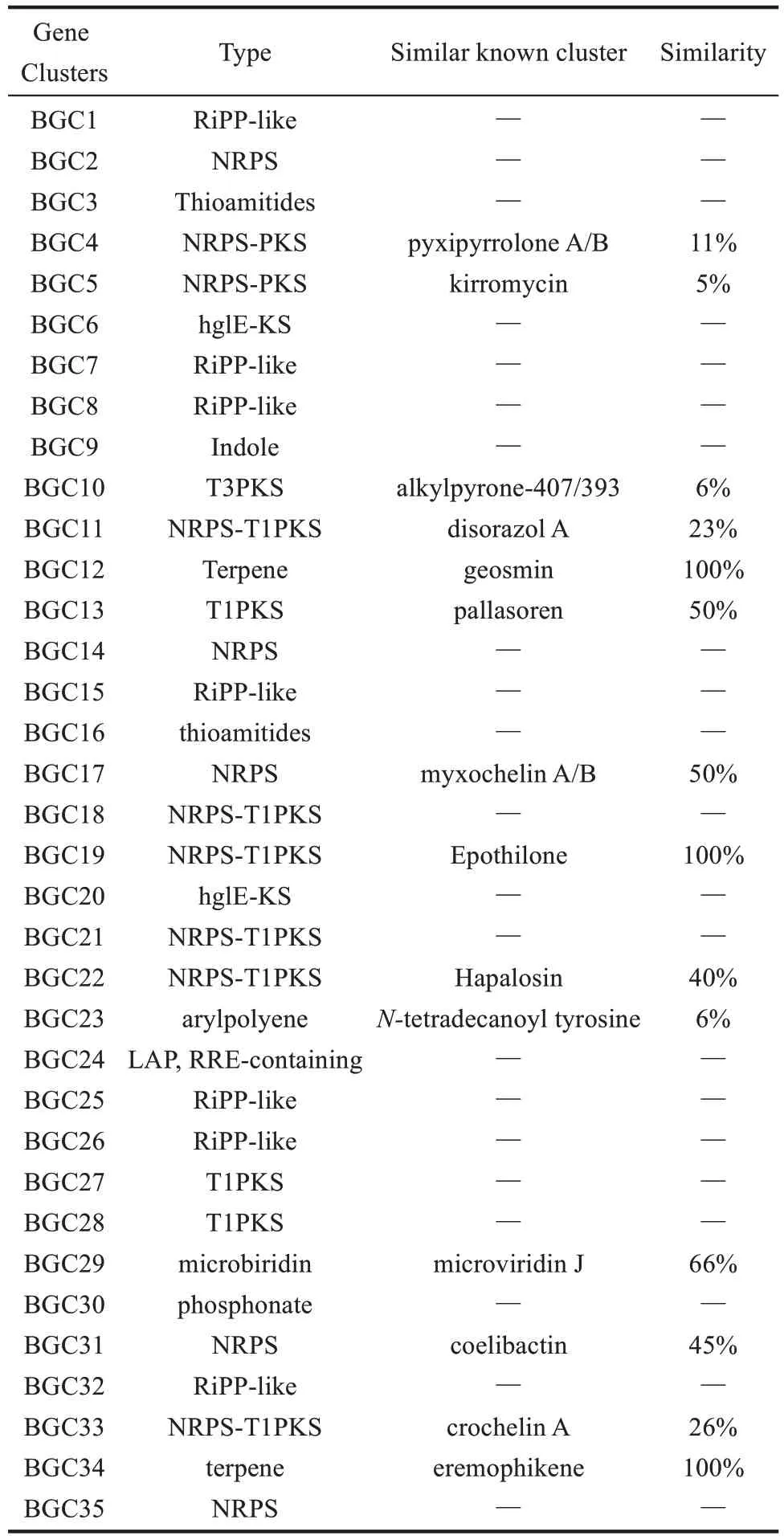
表3 antiSMASH 预测So0157-2基因组编码的生物合成基因簇Tab.3 Biosynthetic gene clusters in the genome of so0157-2 predicted by antiSMASH
2.2 BGC18 的直接克隆、改造、异源表达
利用DraⅠ和Hind Ⅲ酶切基因组,释放出27 kb 的完整BGC18,将酶切后的基因组回收备用。然后PCR扩增1.79 kb的直接克隆载体p15A-cm,得到的片段两端各带有72 bp 的同源臂。参照ExoCET 的方法[22],对基因簇进行克隆(图 2)。复苏后的菌体涂布含氯霉素的LB 筛选平板,过夜培养后挑取24 个转化子,用MscⅠ酶切鉴定(图3),将其中4 个所有酶切条带均正确的质粒,对它们的同源臂部分进行测序,测序正确的命名为p15A-cm-BGC18。为了让质粒p15A-cm-BGC18能在异源宿主中成功表达,需要插入转座元件oriT-tnpA-ⅠR 将基因簇通过转座的方式整合至异源宿主的基因组上。此外,来源于黏细菌的启动子在DSM 7029中可能无法正常工作,所以将BGC18的结构基因的启动子替换成异源宿主中可以工作的组成型启动子Tn5-kan(Pkm)。用带有50 bp 同源臂的引物对pR6K-oriT-TnpA-ⅠR-km 进行扩增,得到oriT-TnpA-ⅠR-km 片段,将这个片段与p15A-cm-BGC18 发生线环重组(LCHR),挑选7 个转化子进行酶切分析和同源臂部分测序,成功获得重组质粒p15A-oriT-ⅠR-Pkm-BGC18(图3)。
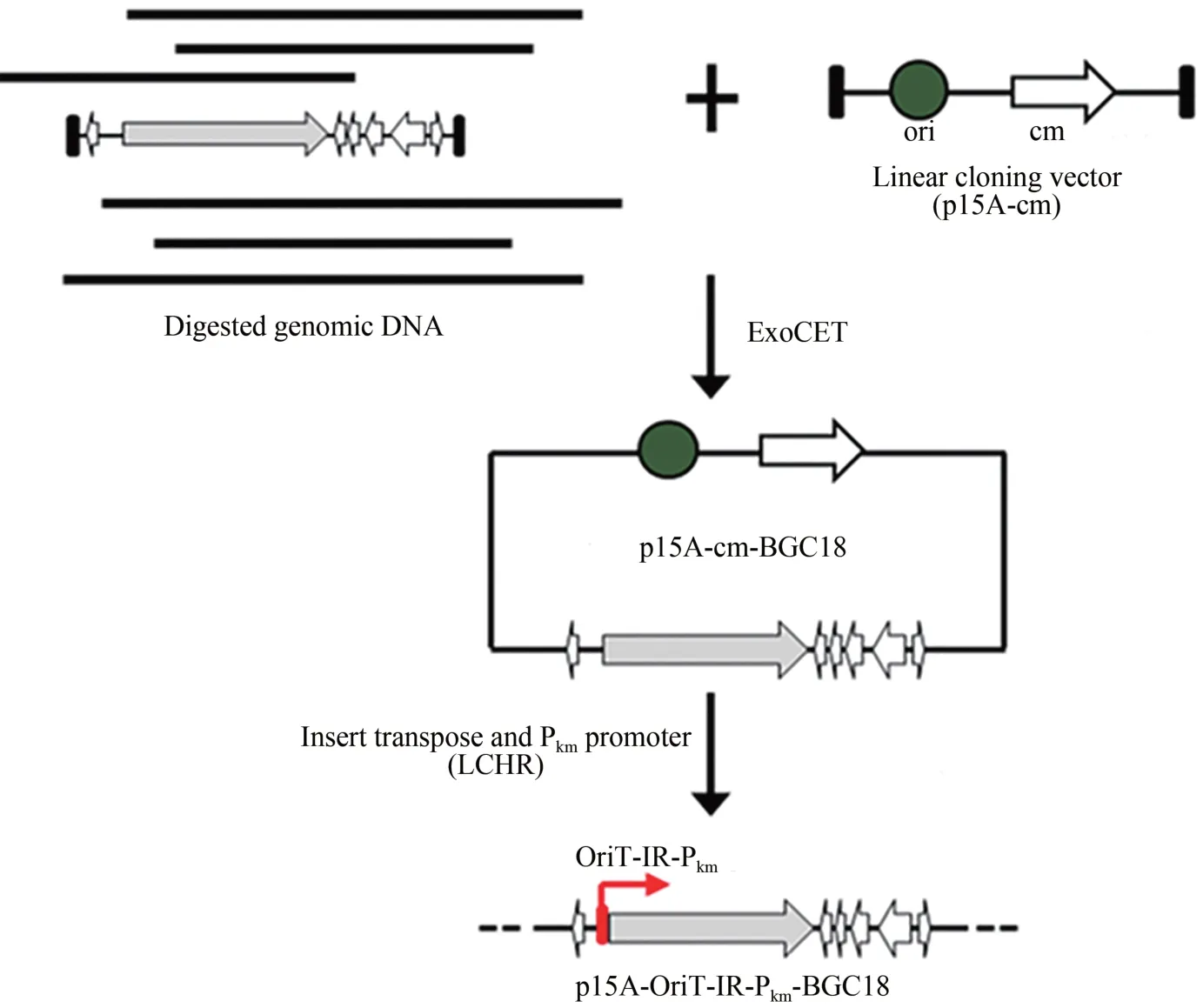
图2 基因簇BGC18的直接克隆与遗传修饰Fig.2 Direct cloning and modification of BGC18
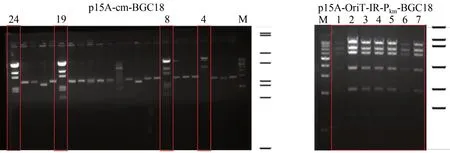
图3 重组质粒 p15A-cm-BGC18(a)和p15A-OriT-ⅠR-Pkm-BGC18(b)分别以MscⅠ和 ApaLⅠ酶切鉴定(红色方框代表酶切条带正确)Fig.3 a:Restriction analysis of the recombinant plasmids p15A-cm-BGC18 by MscⅠ(a)and p15A-OriT-ⅠR-Pkm-BGC18 by ApaLⅠ(b)(Red box indicates right recombinant plasmids)
将质粒 p15A-oriT-ⅠR-Pkm-BGC18 电转入野生型的S. brevitaleaDSM 7029 中,从含卡那霉素的CYMG 平板上挑取重组子进行菌落PCR 鉴定,挑选3 个正确的重组子(DSM7029:Pkm-BGC18)进行发酵检测,并以野生型DSM 7029 作为阴性对照。利用HPLC-MS 对发酵结果进行分析。HPLCMS结果显示,所有带有BGC18的突变体均产生了两个显著的化合物峰m/z227[M+H]+、241[M+H]+,且这些峰在阴性对照中未发现,推测是BGC18 在DSM 7029中表达的产物(图4)。

图4 BGC18在S.brevitalea DSM 7029中的异源表达产物LC-MS分析(BPC+:m/z 200~300)Fig.4 LC-MS analysis for the heterologous products of BGC18 expressed in S.brevitalea DSM 7029
2.3 产物的分离纯化
将突变菌株DSM7029:Pkm-BGC18采用CYMG培养基批量发酵10 L,大孔树脂XAD-16 吸附目标化合物,然后用甲醇提取发酵粗提物。发酵粗提物首先经过正相硅胶柱色谱分离,干法上样,以CH2Cl2-MeOH为流动相,梯度洗脱(100∶1、50∶1、30∶1、20∶1、10∶1),得到 5 个分组分 Fr1~Fr5。液质检测将含有目标化合物的组分Fr3 继续通过反相中压液相色谱制备,水和甲醇为流动相,梯度洗脱。最终通过HPLC 制备,30%乙腈水溶液恒梯度洗脱得到化合物1(33 mg),40%乙腈水溶液恒梯度洗脱得到化合物2(7 mg)和化合物3(4 mg)。
2.4 化合物1-3 结构鉴定
化合物1 为白色固体,阳离子质谱ESⅠMS 在m/z227.0 处给出[M+H]+分子离子峰,结合化合物的一维1H 谱、13C 谱(表4)可以得到化合物分子式为C12H12O2N2。分析13C NMR,DEPT 谱发现,化合物 1 具有 2 个酰胺羰基(δC167.6,165.1);4个次甲基(δC60.1,59.2,32.6,25.1);1个亚甲基 (δC42.7);5 个甲基 (δC32.1,23.1,21.8,19.2,18.0)其中1 个与氮相连;进一步通过HMBC二维核磁数据确定化合物1的结构与已知有机 合 成 中 间 体 Cyclo (N-Me-L-Leu-L-Val) 相同[43-44](图5),但是其核磁数据未见报道,因此对其核磁数据H和C谱进行了解析与指认(表4)。
化合物2 和3 均为白色固体,阳离子质谱ESⅠMS 在m/z241.0 处给出[M+H]+分子离子峰,提示化合物分子式为C13H24O2N2比1 多一个CH2。与化合物1 的氢谱数据比较发现(表4),化合物2中的两个氨基酸均为亮氨酸(Leu)。进一步查阅文献发现化合物2 为已知有机合成中间体Cyclo(N-Me-L-Leu-L-Leu)(图 5)[45]。化合物 3 与化合物2 的氢谱数据非常类似,除了一个二重峰(d)的甲基变成了三重峰(t)的甲基(δH0.84)以及一个与氮相连的CH 信号由dt 峰变为dd 峰(δH3.61),这就表明化合物3中的氨基酸为Ⅰle,为新化合物(图5)。采用Marfey 法对氨基酸的绝对构型进行了确定,4 种氨基酸(N-Me-Leu、Val、Leu、Ⅰle)均为L构型(表5)。
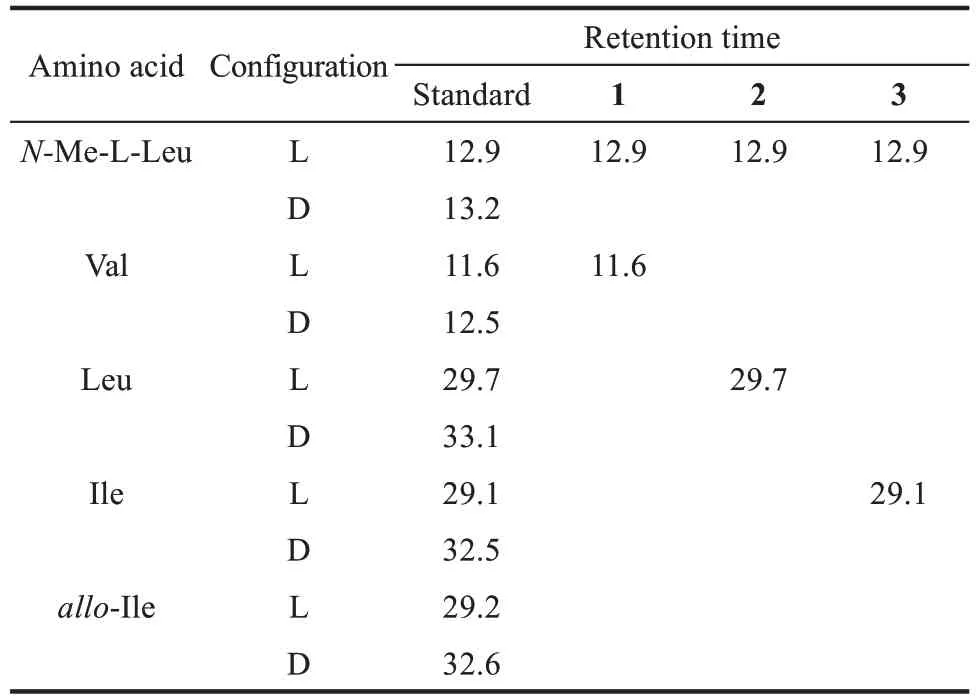
表5 化合物1~3氨基酸与marfey试剂衍生产物的保留时间Tab.5 Retention time of amino acids derivatized with Marfey's reagent

图5 化合物1~3的结构式及化合物1的HMBC相关信号Fig.5 Structures of 1~3 and HMBC correlations of 1
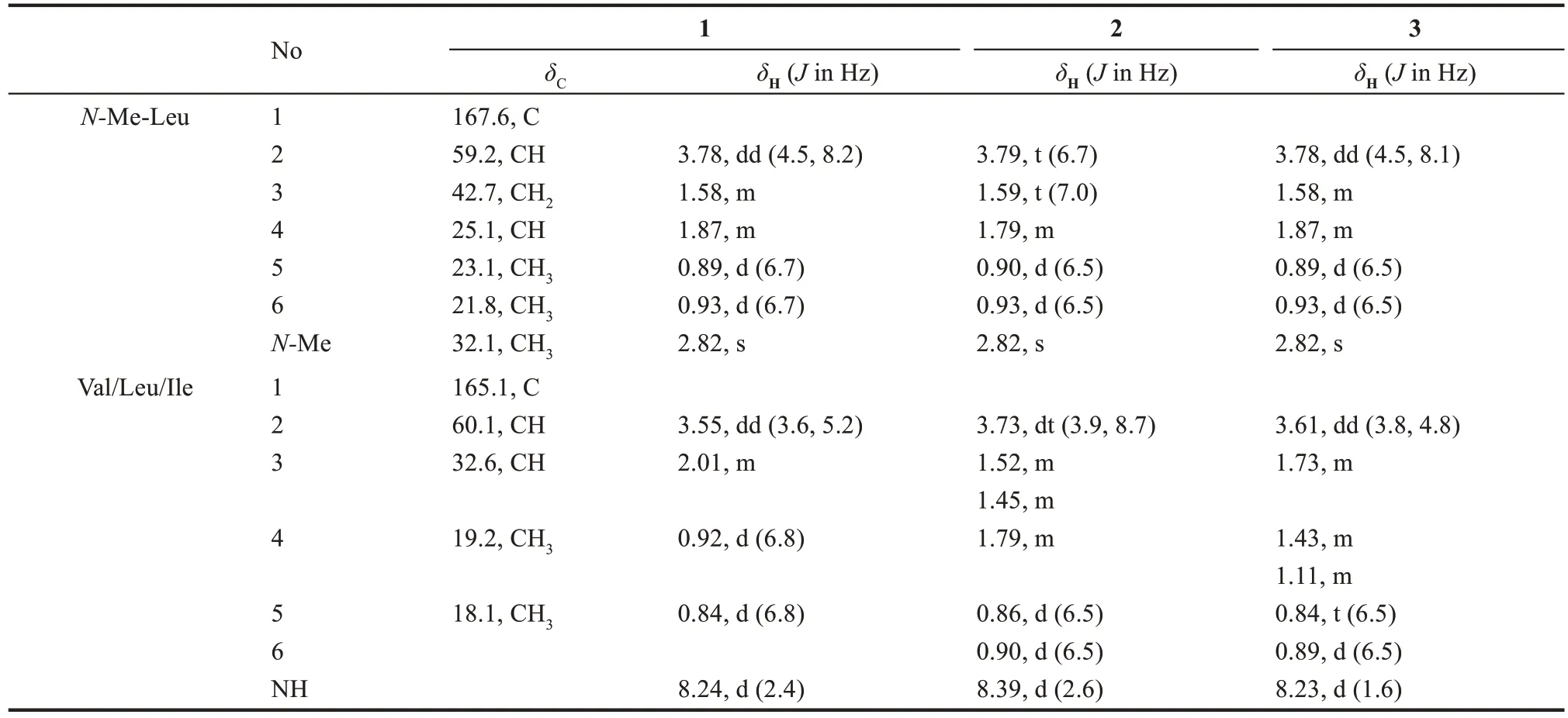
表4 化合物1~3的NMR核磁数据Tab.4 1H(500 MHz)and13C(125 MHz)data of 1~3 in DMSO-d6
2.5 化合物1~3 的生物合成途径分析
根据化合物的结构及其生物合成基因簇的生信分析,对化合物1~3 的生物合成途径进行了推测 (图 6)。A1结构域可识别 L 型的 Val/Leu/Ⅰle。A2结构域可识别的L-Leu,然后经过N-甲基转移酶甲基化修饰后形成N-Me-Leu。两部分经过缩合与TE 介导的环化,最终形成化合物1~3 的完整结构。尽管该基因簇含有PKS 模块,推测可能因为缺少PKS 所必需的T 结构域导致PKS 模块被跳过,从而只获得了NRPS 指导合成的环二肽产物1~3。

图6 化合物1~3生物合成途径推测Fig.6 Proposed biosynthetic pathway of 1~3
2.6 化合物1~3 的抗菌活性测试
本研究采用96孔板二倍稀释法[35]测定了化合物1~3 对下列指示菌株(E. coliATCC 35218、P.aeruginosaATCC 27853、Staphylococcus aureusATCC 29213、Bacillus subtilisATCC 6633、Helicobacter pyloriG27、H.pylori159、Enterococcus faecalisATCC 19434、Mycobacterium smegmatisATCC 607、Candida albicansSC5314)的抗菌活性,结果显示化合物1~3 对所测菌株均无抑制作用(MⅠC>32 μg/mL)。
3 结 论
基因组信息分析表明,纤维堆囊菌So0157-2基因组中含有丰富的未知功能的基因簇,特别是含有大量通常具有良好生物活性的PKS、NRPS 或两者杂合的基因簇,预示着该菌株仍具有产生新颖天然产物的潜力。BGC18 即为该菌基因组中预测的一个NRPS-PKS 杂合的基因簇,大小约为26.7 kb。该基因簇中NRPS 模块的核心基因预测含有两个A 结构域,其中A2结构域被预测具有底物的专一性,而A1结构域对底物的识别具有宽泛性,因此该基因簇可能合成结构多样的二酮哌嗪类化合物。此外,该基因簇PKS模块缺少T结构域,这些底物能否通过NRPS和PKS的延伸单元被依次加载、缩合,形成的终产物最后被TE 结构域释放值得深入研究。然而,由于该菌株较难培养且自身未建立遗传操作体系,因此将该生物合成基因簇转移到合适的异源宿主中,利用异源表达策略是激活该生物合成基因簇挖掘其次级代谢产物的一个有效途径。异源表达成功的关键主要涉及基因簇的克隆与遗传修饰以及异源宿主的合理选择。本课题组已经建立了以Red/ET DNA 同源重组技术为核心的生物合成基因簇直接克隆与遗传修饰技术平台,为天然产物的挖掘提供了技术支撑[21-28]。基因簇在异源表达时,存在着密码子偏好性的影响。选择异源宿主时,首先考虑与原始生产菌在进化地位上亲缘关系相近的菌。本文中所选用的异源宿主S. brevitaleaDSM 7029 与纤维对囊菌So0157-2 均为变形菌门,GC 含量相似,分别为68%和72%[8,39]。前期,本课题组将纤维堆囊菌来源的埃博霉素成功地在该异源宿主S. brevitaleaDSM 7029 中进行了表达,说明两株菌的密码子有一定的兼容性[36]。此外,通过启动子工程改造以及额外补充稀有密码子的tRNA 或者进行密码子优化等也可以提高异源表达的概率[36-37]。
因此,本研究成功构建了纤维堆囊菌So0157-2中基因簇的直接克隆与遗传修饰体系,并将一个NRPS-PKS 杂合的基因簇BGC18 转入异源宿主S.brevitaleaDSM 7029中实现了该基因簇的功能性表达,成功获得了3个对应的代谢产物。通过正相硅胶柱色谱和HPLC 分离纯化,以及MS、NMR 解析了化合物的结构。SciFinder 数据库检索证实化合物1 和2 仅作为有机合成中间体被报道,为新天然产物,而化合物3为新化合物。BGC18为NRPSPKS 杂合基因簇,从结构上看,化合物1~3 为二酮哌嗪类化合物,其所含氨基酸与生物信息学预测一致,但是并未见PKS 单元。仔细分析PKS 相关基因发现该基因簇缺少硫醇化结构域,推测导致PKS不能有效组装。化合物1~3结构上差别主要为第1 个氨基酸的不同(1 Val, 2 Leu, 3 Ⅰle),这是由于第1 个腺苷化结构域(A domain)对底物识别的非特异性所导致的。由A 结构域对底物识别的宽泛性导致的结构多样性也在我们前期分离获得的多个脂肽类化合物得到证实[46-47]。基于Red/ET 重组工程技术的纤维堆囊菌So0157-2 隐性基因簇的直接克隆、修饰和异源表达体系的建立,不仅有助于进一步了解该菌的生物合成潜力,而且能够发现新颖的天然产物用于药物筛选评价。
致谢:本工作得到国家重点研发计划(2019YFA0905 700);国家自然科学基金(32070060,31670098);山东省自然科学基金(ZR2020QH345,ZR2019JQ11)的支持。
