家族性高胆固醇血症研究进展
2021-11-27蒋琬姿张丽雯贺彩红阮梅花季勇于建荣周红文
蒋琬姿,张丽雯,贺彩红,3,阮梅花,季勇,于建荣,3,周红文
领域动态
家族性高胆固醇血症研究进展
蒋琬姿1,张丽雯2,贺彩红2,3,阮梅花2,季勇4,于建荣2,3,周红文1
1. 南京医科大学第一附属医院内分泌科,南京 210029 2. 中国科学院上海生命科学信息中心,中国科学院上海营养与健康研究所,上海 200031 3. 中国科学院大学,北京 100049 4. 南京医科大学,南京 210029
家族性高胆固醇血症(familial hypercholesterolemia, FH)是以肌腱黄瘤、低密度脂蛋白胆固醇(low density lipoprotein cholesterol, LDL-C)显著升高和早发冠心病(premature coronary artery disease, PCAD)为特征的一种常染色体显性或隐性遗传病。本文分析了FH的国内外研究现状,总结了目前中国已报道的FH人群相关的基因突变位点及治疗现状,同时统计了FH相关专利及药物研发情况。在论文发表方面,FH致病机制与治疗、未成年FH患者研究等成为研究热点;在专利方面,再生元制药、阿斯利康、默克等大型药企在FH检测、诊断、治疗等方面积极探索;在药物研发方面,已有12种药物在美国、日本、欧洲等国家/地区上市,为FH患者带来希望。
家族性高胆固醇血症;罕见病;FH基因突变位点;研究现状
家族性高胆固醇血症(familial hypercholesterolemia, FH)是以肌腱黄瘤、低密度脂蛋白胆固醇(low density lipoprotein cholesterol, LDL-C)显著升高和早发冠心病(premature coronary artery disease, PCAD)为特征的一种常染色体遗传性疾病[1]。临床上分为杂合子FH(HeFH)和纯合子FH(HoFH),杂合子及纯合子患者血浆LDL-C水平分别为正常人的2~3倍和6~8倍[2]。流行病学研究表明,全球FH的总体患病率约为1/500,FH纯合子患病率约为1/100万[3];近年来也有研究表明,FH的总体发病率为1/200~1/500,FH纯合子预估患病率为1/30万~1/60万[4~6]。但是世界上绝大部分国家和地区FH患者诊断率仍然小于1%[7],且治疗状况差,大部分患者未达到指南推荐的LDL-C目标水平[8]。中国作为世界上人口最多的国家,FH患者约占全球所有FH患者的8%[9]。协和医院张抒扬教授依据中国总人口预估的纯合子FH患病人数约为2205~4609例,杂合子患病人数约为276万~691万[9]。
FH主要为常染色体显性遗传,部分为常染色体隐性遗传。已鉴定出的致FH突变基因中,显性遗传基因主要包括低密度脂蛋白受体(low density lipoprotein receptor,)、前蛋白转化酶枯草溶菌素9 (proprotein convertase subtilisin/kexin type 9,)及载脂蛋白B (apolipoprotein B,)基因[10];隐性遗传基因则主要为LDLR衔接因子蛋白1 (low-density lipoprotein receptor adapter protein 1,)基因[11]。近年来也有文献报道FH致病新基因,如环氧化物水解酶2 (epoxide hydrolase 2,)、生长激素受体(growth hormone receptor,)、载脂蛋白E (apolipoprotein E,)基因等[12]。
《家族性高胆固醇血症筛查与诊治中国专家共识》[13]指出,成人符合下列标准中的2项即可诊断为FH:(1)未经调脂药物治疗的患者血清LDL-C水平≥4.7 mmol/L (180 mg/dL);(2)有皮肤/腱黄色瘤或<45岁的人存在脂性角膜弓;(3)一级亲属中有FH或早发动脉粥样硬化性心血管疾病,特别是冠心病患者。儿童FH的诊断标准:未治疗的血LDL-C水平≥3.6 mmol/L (140 mg/dL)且一级亲属中有FH患者或早发冠心病患者。检测出、、和基因致病性突变也可诊断为FH。本文从FH基因突变位点、治疗现状、专利、药物研发等方面分析FH的总体研究现状,为我国FH的研究与治疗提供参考。
1 FH主要突变基因
、、、及基因的突变大多数是由外显子区域碱基替换及小片段缺失导致,本文将进一步介绍、、、及基因的突变位点研究现状(截至2020年中国FH患者主要突变基因相关情况详见附表1~4)。
1.1 LDLR
基因位于染色体19p13.1-13.3,包含18个外显子和17个非编码区,大约89% FH患者为基因突变[14]。LDLR主要位于肝细胞膜表面,该基因突变可导致LDLR的功能和活性发生变化,无法清除外周血中的LDL-C,从而导致血LDL-C浓度升高[5]。
据LOVD数据库(https://databases.lovd.nl/shared/ genes/LDLR),截至2020年9月,全球共有3731个基因突变的相关文献报道,涉及2034个突变位点,且有1364种突变仅见一次报道。在突变中,约70%为碱基置换突变,20%为缺失突变,其余为插入突变、复制突变、反义突变等。在18个外显子中,第4外显子(exon 4, E4)的突变占有较大比例,约为20%,可能原因为E4在染色体上的跨度最大、所含碱基最多及选择偏倚;E15和E16的突变率相对较低。
FH人群中主要突变形式有:c.2177C> T、c.1775G>A、c.2054C>T、c.1048C>T、c.682G>A、c.829G>A、c.1285G>A、c.798T>A、c.(940+1_941-1)_ (1186+1_1187-1)del、c.1567G>A、c.530C>T、c.1222G>A、c.1432G>A等。
据统计[15],1950~2019年,中国大陆、中国香港、中国澳门地区共报道177个突变,主要包括:c.1747C>T、c.1448G>A、c.1879G>A、c.313+1G> A、c.2054C>T、c.986G>A、c.1765G>A、c.1879G> A、c.1432G>A等,其中E10上c.1448G>A (第1448位碱基G被A取代)为最常见突变,其氨基酸改变为p.Trp483X (第483位色氨酸被终止密码子取代);中国台湾地区共报道81个突变,主要包括:c.1747C>T、c.986G>A、c.268G>A、c.1322T>C、c.1432G>A、c.1246C>T等,其中E12上c.1747C>T (第1747位碱基C被T取代)为最常见突变,其氨基酸改变为p.His583Tyr (第583位组氨酸被酪氨酸取代)。
1.2 Apo B
基因位于染色体2p24.1,该基因主要编码LDL中的载脂蛋白B-100 (ApoB-100),是与LDLR结合的主要配体,大约9% FH患者为该基因突变[14]。基因突变可导致ApoB-100的LDLR结合域发生改变,导致载有脂质的LDL无法与肝细胞膜表面的 LDLR正确结合,从而导致血中LDL清除减少,血LDL-C浓度升高[16]。
截至2020年9月,LOVD数据库共记录有全球1148个基因突变的相关文献报道,涉及958个突变位点,且有759种突变仅见一次报道。在突变中,约90%为碱基置换突变,8%为缺失突变,2%为插入及复制突变。
FH人群中主要突变形式有:c.10579C> T、c.10580G>A、c.10913G>A、c.7696G>A、c.5741A> G、c.13444A>G、c.4838G>C、c.12382G>A、c.10294C> G、c.3383G>A、c.293C>T等。
亚洲人群中突变的主要形式为p. Arg3527Trp。1950~2019年,中国大陆、中国香港与中国澳门地区FH人群共报道47个突变,其突变形式有:c.10579C>T、c.1594C>T、c.6110T> C、c.7223C>T、c.8267G>T、c.8462C>T、c.889C>T、c.9164A>G、c.2870T>C、c.10748A>T、c.581C>T、c.11052A>T、c.11585T>C,其中以c.10579C>T及c.11585T>C突变较为常见[12,17~21];中国台湾地区共报道58个突变,较常见的突变形式有c.10707C>T、c.10579C>T、c.10828 C>T[22~24]。
1.3 PCSK9
基因位于染色体1p32.3,有12个外显子,属于前蛋白转换酶家族[10],大约2% FH患者为该基因突变[16]。PCSK9可与肝细胞表面LDLR的表皮生长因子A (epidermal growth factor-A, EGF-A)结构域结合,通过增加LDLR内吞和溶酶体降解作用来减少肝脏LDL-C的吸收,进而导致血LDL-C升高[25]。
截至2020年9月,LOVD数据库共记录有全球286个基因突变的相关文献报道,涉及258个突变位点,且有233种突变仅见一次报道。在突变中,约87%为碱基置换突变,8%为缺失突变,其余为插入突变、复制突变。
根据突变对PCSK9功能的影响,可分为功能获得型和功能缺失型突变两种,前者导致胆固醇水平升高,FH人群中主要突变形式有:c.1487G> A、c.1120G>T、c.10G>A、c.94G>A、c.1486C>T、c.1405C>T、c.658G>A、c.277C>T、c.7454A>G等[26]。
亚洲人群中最常见的突变为c.94G>A;中国FH人群中主要的突变形式有:c.277C> T、c.1792G>A、c.10G>A、c.644G>A、c.626C>T、c.63-65insCTG、c.287G>T、c.313C>T、c.918G>T等,研究表明c.287G>T及c.313C>T错义突变在中国FH人群中最为常见[19]。
1.4 LDLRAP1
基因位于染色体1p36.11,由9个外显子组成。该基因编码308个氨基酸,LDLRAP1与LDLR可结合成LDL-LDLR复合物进行内吞并输运到胞内[27]突变导致LDLRAP1蛋白的分子缺陷,严重减少LDL-C摄取,从而降低LDL-C的代谢。
截至2020年9月,据LOVD数据库,全球共有88个基因突变的相关文献报道,涉及86个突变位点,且有84种突变仅见一次报道。在突变中,约91%为碱基置换突变,6%为缺失突变,3%为复制突变。
主要突变形式有:c.396C>T、c.654A> G、c.65G>A、c.71dup、c.71G>T、c.223G>A、c.284G> A、c.397G>A、c.406C>T、c.413A>G、c.423C>T等。
1.5 EPHX2
基因位于染色体8p21.2-p21.1,包含19个外显子,编码555个氨基酸,是α/β-水解酶家族成员[28]。EPHX2广泛存在于哺乳动物组织中,在自然界中其N端磷酸酶结构域及C端水解酶结构域以二聚体形式稳定存在[29]。其中N端具有去磷酸化作用可调节胆固醇水平和信号传递,而C端可催化花生四烯酸类环氧化合物(epoxyeicosatrienoic acids, EETs)水解产生二醇类物质[30]。通过EETs和其他脂质介质的代谢,可溶性环氧化物水解酶在高血压、心脏肥大、动脉硬化、脑和心脏缺血/再灌注损伤等多种疾病中发挥作用。突变降低细胞LDLR结合及内吞能力,从而使得血LDL-C浓度升高[28]。
截至2020年9月,根据LOVD数据库,全球共有6个基因突变的相关文献报道,涉及6个突变位点,均仅见一次报道。在突变中,约66.7%为碱基置换突变,33.3%为缺失突变。
主要突变形式有:c.-86G>C、c.-5G> A、c.230G>A、c.313A>G、c.502-19A>G、c.577-3C>T。
1.6 GHR
基因位于染色体5p13.1-p12,包含10个外显子,编码620个氨基酸,是属于细胞因子受体超家族的单一跨膜糖蛋白[31]。生长激素与跨膜受体GHR结合,激活的GHR通过细胞内信号转导途径介导细胞反应,导致胰岛素样生长因子的合成和分泌。突变可导致莱伦氏综合征,临床表现有生长迟缓、骨龄延迟、马鞍鼻、声音音调高亢、男性生殖器较小、骨质疏松、肌肉发育不良、运动能力发展迟缓等。代谢特点是空腹低血糖,至少50%的婴儿和儿童有过明显的低血糖症状,另可伴有血LDL-C水平升高。有研究表明,突变导致GH上调肝脏LDLR mRNA表达、加速LDL-C分解代谢和降低组织的脂质含量刺激LDL-C清除等作用减弱[32]。
截至2020年9月,据LOVD数据库,全球共有140个基因突变的相关文献报道,涉及78个突变位点,且有39种突变仅见一次报道。在突变中,约86%为碱基置换突变,9%为缺失突变,其余为插入突变、复制突变。
主要突变形式有:c.181C>T、c.558A> G、c.703C>T、c.1483C>A、c.1735C>A等。
1.7 Apo E
基因位于染色体19q13.32,编码317个氨基酸的Apo E前体,18个氨基酸的信号肽裂解和糖基化后,成熟的Apo E以299个氨基酸的蛋白质分泌[33]。Apo E作为脂蛋白的结构蛋白,构成脂蛋白的外层,主要分布于极低密度脂蛋白、高密度脂蛋白、乳糜微粒中,作为LDLR家族的配体,调节脂质代谢等多种生物学反应。
截至2020年9月,据LOVD数据库,全球共有240个基因突变的相关文献报道,涉及88个突变位点,且有72种突变仅见一次报道。在突变中,约86%为碱基置换突变,9%为缺失突变,其余为插入突变、复制突变。
主要突变形式有:c.388T>C、c.526C> T、c.942C>T、c.90C>G、c.149G>A等。
2 研究趋势
2.1 致病机制与疾病治疗为FH研究热点
在Web of Science核心合集数据库中1检索时间:2021-03-16,数据库更新时间:2021-03-15,文献类型:Article+Review,在主题中检索,检索式为:TS = ("familial hypercholesterolemia" or "Familial hypercholesterolaemia" or "familial hyperbetalipoproteinaemia" or "familial hypercholesteremia" or "Fredrickson type IIa hyperlipoproteinemia" or "Fredrickson type IIa lipidaemia" or "Hyperlipoproteinemia type IIA" or "Type II Hyperlipidemia")检索FH相关论文,得到论文8676篇(截至2020年)。选取2016~2020年的2159篇论文对研究热点、主要国家/地区进行分析。

图1 FH论文年度趋势
Web of Science核心合集数据库收录的最早的FH相关论文始于1948年。1974年,Brown等[34]在杂志上首次发文报道了基因突变可导致FH,推动了FH相关发病机制的研究(图1),Brown也因此获得了1985年的诺贝尔生理学及医学奖。2003年,Abifadel等[10]通过对基因的12个外显子进行测序,首先报道了新的基因突变位点,揭示了一种新的致FH机制,促进了FH领域研究的发展。
在论文发表方面,2016~2020年,FH领域发文量排名前10位国家/地区分别为:美国、英国、意大利、加拿大、中国、荷兰、澳大利亚、德国、法国、日本。从年度趋势来看,2016~2019年中国论文发表量不断增长,随着发文量上升,于2019年超过英国、意大利等居第二位,而2020年发文量较2019年出现一定程度下降。使用VOSviewer软件进行关键词聚类分析,可以看出,FH的研究重点主要围绕基因表达与疾病发生、流行病学研究、临床试验与治疗、未成年人家族性高胆固醇血症疾病发生等四方面展开(图2)。
(1)基因表达与疾病发生(红色):包括等致病基因的表达、变异与疾病的关系及相关诊断方式、技术等。
(2)流行病学研究(绿色):主要对FH等心血管疾病的发病率、危险因素、临床症状、预防与控制等展开研究。
(3)临床试验与治疗(蓝色):主要包括FH的等主要致病基因抑制剂的临床试验及相关抗体、药物的制备等。
(4)未成年人家族性高胆固醇血症疾病发生(黄色):主要对青少年、儿童等未成年人FH等心血管疾病发生、治疗及防控手段进行研究。
2.2 诊断治疗成为FH专利技术研发重点
在Incopat数据2检索日期:2021-01-08中检索FH相关专利,共得到相关专利541件,并对这些专利进行深入分析。
FH领域公开专利最早可追溯到2002年,2002~2004年,专利申请数有较大程度增长,到2014年,申请与公开专利数量均呈波动下降趋势。2014~ 2018年,FH相关专利申请与公开数量上升;2018年后专利公开数量呈波动上升趋势(图3)。
对FH专利主题词进行聚类分析,主要可分以下四类。
(1) FH基因疗法及他汀外药物降脂活性剂研究:涉及ANGPTL8抑制剂、ANGPTL3抑制剂、内皮脂肪酶抑制剂、PCSK9抑制剂等药物研发及FH的基因疗法研究。

图2 2016~2020年FH领域研究热点分析

图3 FH领域专利申请与专利公开年度趋势
由于专利申请后评审需要18个月后才能公开,因此2019和2020年的专利量不全,仅供参考。
(2) FH他汀类药物研发与应用:主要包括瑞舒伐他汀、阿托伐他汀、辛伐他汀等HMG-CoA还原酶抑制剂类药物的研究。
(3) FH检测/诊断/预后方法研究:主要涉及基因突变检测方法与试剂盒开发及FH易感性相关SNP位点的引物、检测方法研究、FH的体外诊断方法、饮食辅助疗法预后等研究。
(4) FH片剂/胶囊剂/粉剂药物制备方法与优化:主要为辛伐他汀胶囊剂、匹伐他汀钙片剂、考来替兰分散片、奥美沙坦酯/瑞舒伐他汀复方制剂、阿托伐他汀钙化合物等药物制备及优化(如添加碱性金属增强稳定性)方法研究。
对FH领域全球Top5专利权人(美国再生元制药、美国宾夕法尼亚大学、英国/瑞典阿斯利康、美国默克、美国德克萨斯大学)的专利布局进行分析,前5专利权人申请的专利集中在FH检测、诊断及治疗方面,又各有侧重。再生元制药申请的专利主要围绕PCSK9抑制剂、ANGPTL8抑制剂、ANGPTL3抑制剂等药物的研发;宾夕法尼亚大学重点围绕FH诊断方法及基因突变检测方法进行布局;阿斯利康与默克专利布局主要集中在瑞舒伐他汀等他汀类药物研发应用方面;德克萨斯大学则主要围绕FH的检测方法进行布局。
3 治疗进展
由于FH患者出生后即处于高LDL-C水平暴露状态,其罹患动脉粥样硬化性心血管疾病(arteriosclerotic cardiovascular disease, ASCVD)风险明显增高。因此尽早开展级联筛查,早期诊断和早期治疗是改善FH患者临床预后的重要措施。我国FH患者的治疗目标如下:合并与不合并ASCVD的成人FH患者血LDL-C的目标值分别为<1.8 mmol/L (70 mg/dL)和<2.6 mmol/L (100 mg/dL);儿童FH患者血LDL-C的目标值<3.4 mmol/L (130 mg/dL)。若难以达到上述目标值,建议至少将血清LDL-C水平降低50%[13]。
目前针对FH患者的降脂治疗的主要方式包括改善生活方式、药物干预治疗、脂蛋白血浆置换及肝脏移植等。
3.1 改善生活方式
健康科学的生活方式是FH治疗的基础措施,鼓励患者戒烟,坚持低饱和脂肪酸、低胆固醇饮食。控制体重,建议患者积极参加体育锻炼(在体育活动开始之前仔细评估心血管风险)[13]。
3.2 药物干预治疗
FH诊断后应立即启动降胆固醇药物治疗[13]。
3.2.1 全球已有12个FH药物上市
在Cortellis数据库3检索日期:2021-01-25中检索FH相关药物,共获得有效药物23种(包括已上市、已注册、预注册、临床3期、临床2期、临床1期、临床阶段、发现阶段等药物)。
其中,已上市药物12种,分别为:米泊美生(mipomersen)、依折麦布–瑞舒伐他汀钙片(ezetimibe- rosuvastatin calcium)、依折麦布–阿托伐他汀(ezetimibe-atorvastatin)、依折麦布–辛伐他汀(ezetimibe- simvastatin)、依洛尤单抗(evolocumab)、匹伐他汀(pitavastatin)、依折麦布(ezetimibe)、氟伐他汀(口服,缓释型) (fluvastatin, oral, extended-release)、阿托伐他汀(atorvastatin)、阿利珠单抗(alirocumab)、瑞舒伐他汀(rosuvastatin)、洛美他派(lomitapide);处于临床3期的药物有2种,分别为LIB-003、resmetirom。
3.2.2 HMG-CoA还原酶和PCSK9等为FH药物主要靶标
目前,治疗FH的药物主要是他汀类,可联合胆固醇吸收抑制剂或胆汁酸螯合剂,降低LDL-C水平。多种新型降胆固醇药物已被批准用于FH的治疗,包括HMG-CoA还原酶抑制剂(7个)、PCSK9抑制剂(5个)、白细胞介素-1β受体抑制剂(2个)、尼曼-匹克C1型类似蛋白1抑制剂(2个)、微粒体甘油三酯转运蛋白(microsomal triglyceride transfer protein, MTTP)抑制剂(1个)、Apo B合成抑制剂等(1个)、血管生成素样3 (angiopioetin-like protein 3, ANGPTL3)抑制剂(1个)等,其主要作用见表1。
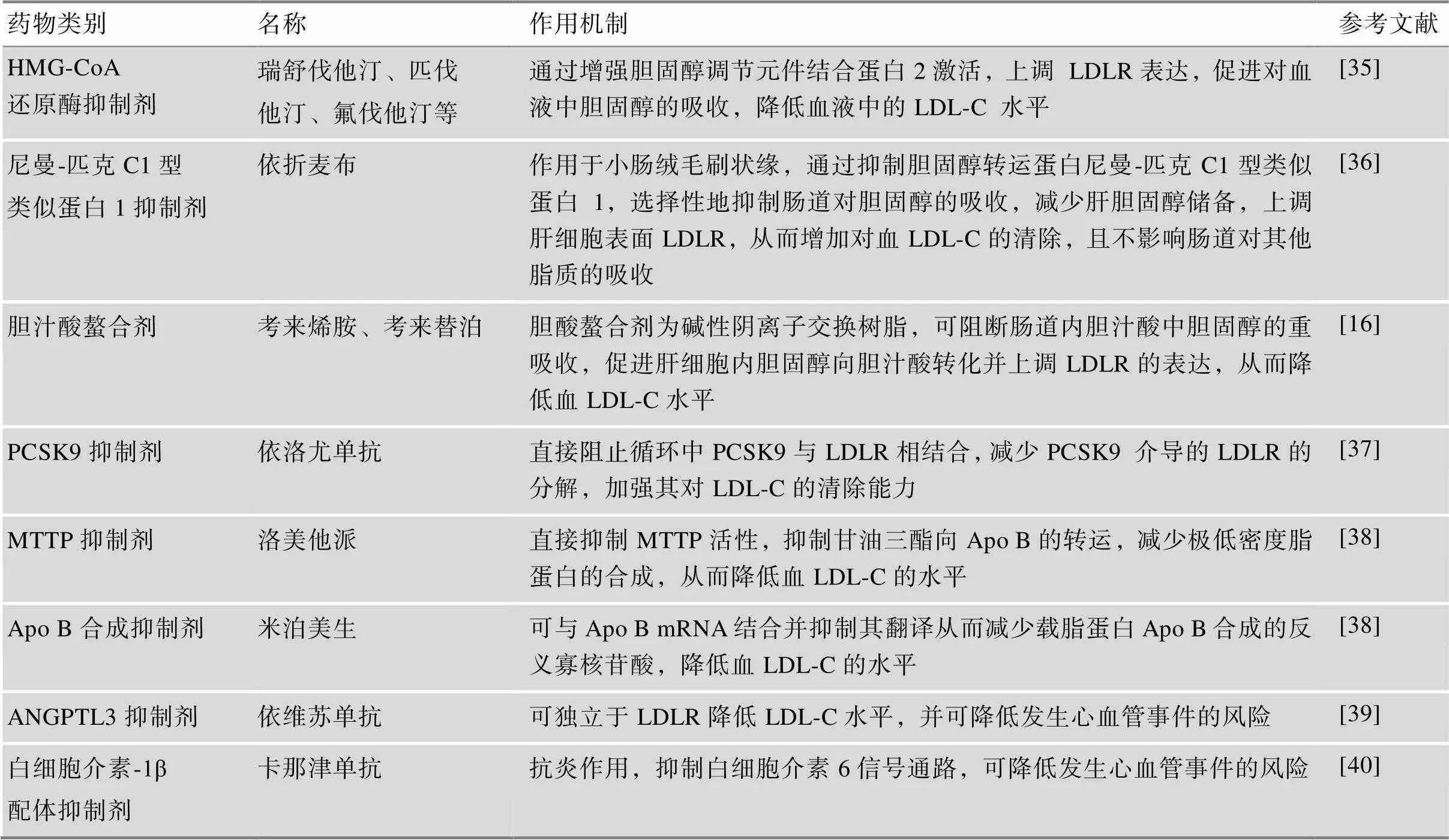
表1 不同类别药物在降LDL-C或抗炎方面的作用
他汀类药物是治疗FH的一线药物。LDLR具有50%以上功能的HeFH患者对他汀类药物反应良好,但HoFH患者对他汀反应较差。治疗效果可能与种族有关,中国患者接受常规剂量他汀类药物治疗反应不佳,对大剂量他汀类药物的耐受性普遍差于西方人[41]。
FH患者除他汀类药物治疗外,还应给予降脂药物,包括依折麦布、胆酸螯合剂、烟酸、洛美他派、米泊美生、PCSK9抑制剂等。但目前洛美他派及米泊美生在中国暂未上市,国内获批上市的药物只有他汀类、依折麦布、PCSK9抑制剂和烟酸。其他药物缺乏针对中国人群的安全性和有效性数据,在上市前仍处于研发或II期和III期临床试验阶段[9]。此外,当烟酸和他汀类药物一起服用时,肌肉毒性的风险会增加。因此,大多数中国FH患者使用他汀类药物联合依折麦布降低血脂水平[42]。研究表明,近年来开发的新型治疗药物PCSK9抑制剂与他汀类药物联合依折麦布可以更好地降低LDL-C水平[43]。2018年8月,依洛尤单抗成为国内首个用于治疗HoFH成人或12岁以上青少年的PCSK9抑制剂,从而推动了中国FH患者治疗新方法的开发,为患者带来希望[9]。
3.2.3 默克、拜耳、诺华等大型制药企业引领FH药物研发
FH领域在研药物数量最多的为美国默克,有3个,分别为依折麦布–瑞舒伐他汀钙片、依折麦布–辛伐他汀、依折麦布,均已上市,主要为HMG-CoA还原酶抑制剂及尼曼–匹克C1型类似蛋白1抑制剂类药物;其次为德国拜耳2个(依折麦布–阿伐他汀、依折麦布–瑞舒伐他汀钙片,均已上市)、美国诺华制药2个(其中,氟伐他汀(口服,缓释型)已上市)、美国再生元制药2个(其中,阿利珠单抗已上市)、日本安斯泰来制药1个(依洛尤单抗,已上市)等。
3.3 脂蛋白血浆置换
若患者药物联合治疗效果欠佳,可考虑脂蛋白血浆置换。血浆置换为有创治疗且费用昂贵,需每1~2周进行1次维持治疗[44],主要适用于HoFH患者,对伴有冠心病的高危HeFH患者或对他汀类药物不耐受或药物治疗下血LDL-C水平仍较高的HeFH患者。有研究表明,脂蛋白血浆置换后,LDL-C和Lp(a)水平可显著降低50%~70%[45]。脂蛋白血浆置换与降脂药物联合使用,可能会进一步改善血脂水平,降低心血管风险;最常见的不良反应是轻度至重度低血压和恶心[45]。
3.4 肝脏移植
肝脏作为清除血胆固醇的主要器官,约80%的LDLR位于肝细胞中,肝移植成为可选治疗方案[46]。通过肝移植纠正肝细胞上、、等基因的分子缺陷,虽然可以降低LDL-C水平,但由于移植后手术并发症和死亡率高以及供体匮乏等因素,难以作为主要的FH治疗手段[13]。
4 结语与展望
近年来,FH的发病率逐年上升,且有年轻化的趋势,FH的就医率及FH的社会认知度处于较低水平。中国FH患者人数预计将达到276万~691万,但诊治率不足1%,从而导致高血脂患者早期心血管事件的发生极高。但对于FH并发冠心病患者,未治疗率也高达20.6%,即便经低、中、高强度降脂治疗(治疗率分别为6.0%、68.3%、5.0%),患者的LDL-C水平也均未达到≤2.6 mmol/L[47]。
在FH患者治疗方面,国内外指南推荐使用他汀类药物,而他汀可能存在不耐受以及肌痛、痉挛、肌酸激酶升高等药物相关肌肉症状,临床迫切需要一种安全性好且能有效降低LDL-C水平的新药物及新疗法。随着基因检测与基因编辑技术、精准医学和个性化医疗的发展,大数据与人工智能在医疗中的广泛应用,FH领域将进一步快速发展。随着对、、、等主要致FH基因及、等新发现致病基因的研究,相应突变基因靶向药将拥有巨大市场。如大量研究证明,PCSK9单克隆抗体不仅能够降低FH患者LDL-C水平、耐受性良好,且具有额外的心血管获益,可作为我国FH患者治疗新选择,助力我国FH的防治。
值得注意的是,由于FH治疗方案的信息大多来自国外研究,迫切需要国内FH患者的临床研究数据,以便在国家层面进一步开展治疗。此外,中国FH儿童长期服用降脂药物的安全性和耐受性仍需进一步关注。
同时,优化FH的诊疗及随访体系,未来可建立FH患者国家电子档案数据库,与各临床医疗中心的电子病历系统对接,对早期筛查、整合与分析患者信息、指导治疗方案、评估治疗效果、实现长期监管、进一步制定适合中国FH患者的诊断标准及治疗方案、改善FH患者远期预后具有重要意义,甚至可为国内外专家的沟通与交流提供支持。同时,在中国人群中开展FH新致病基因的筛选,也具有重要临床与科研价值。
通过健康教育、饮食控制、改善生活方式以及有规律的运动,可有效调节机体脂质以及脂蛋白代谢,实现对FH及相关心脑血管疾病的早期防治。通过基因诊断,可实现FH的早发现、早诊断,未来可结合基因编辑个体化医疗,有望治愈越来越多的罕见病,大幅度提高患者的生活质量。
附加材料详见文章电子版www.chinagene.cn。
[1] Krogh HW, Mundal L, Holven KB, Retterstol K. Patients with familial hypercholesterolaemia are characterized by presence of cardiovascular disease at the time of death., 2016, 37(17): 1398–1405.
[2] Li S, Zhang Y, Zhu CG, Guo YL, Wu NQ, Gao Y, Qing P, Li XL, Sun J, Liu G, Dong Q, Xu RX, Cui CJ, Li JJ. Identification of familial hypercholesterolemia in patients with myocardial infarction: A Chinese cohort study., 2016, 10(6): 1344–1352.
[3] Defesche JC, Gidding SS, Harada-Shiba M, Hegele RA, Santos RD, Wierzbicki AS. Familial hypercholesterolaemia., 2017, 3: 17093.
[4] Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, Wiklund O, Hegele RA, Raal FJ, Defesche JC, Wiegman A, Santos RD, Watts GF, Parhofer KG, Hovingh GK, Kovanen PT, Boileau C, Averna M, Borén J, Bruckert E, Catapano AL, Kuivenhoven JA, Pajukanta P, Ray K, Stalenhoef AFH, Stroes E, Taskinen MR, Tybjaerg-Hansen A, European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society., 2013, 34(45): 3478–3490a.
[5] Gidding SS, Champagne MA, de Ferranti SD, Defesche J, Ito MK, Knowles JW, McCrindle B, Raal F, Rader D, Santos RD, Lopes-Virella M, Watts GF, Wierzbicki AS, American Heart Association Atherosclerosis, Hypertension, and Obesity in Young Committee of Council on Cardiovascular Disease in Young, Council on Cardiovascular and Stroke Nursing, Council on Functional Genomics and Translational Biology, and Council on Lifestyle and Cardiometabolic Health. The agenda for familial hypercholesterolemia: a scientific statement from the American heart Association., 2015, 132(22): 2167–2192.
[6] Cuchel M, Bruckert E, Ginsberg HN, Raal FJ, Santos RD, Hegele RA, Kuivenhoven JA, Nordestgaard BG, Descamps OS, Steinhagen-Thiessen E, Tybjaerg-Hansen A, Watts GF, Averna M, Boileau C, Borén J, Catapano AL, Defesche JC, Hovingh GK, Humphries SE, Kovanen PT, Masana L, Pajukanta P, Parhofer KG, Ray KK, Stalenhoef AFH, Stroes E, Taskinen MR, Wiegman A, Wiklund O, Chapman MJ, European Atherosclerosis Society Consensus Panel on Familial Hypercholesterolaemia. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society., 2014, 35(32): 2146–2157.
[7] Ding YM, Ding YN, Sun WQ, Liu JG. Talking about chronic disease management from the diagnosis and treatment status of patients with familial hypercholesterolemia in China., 2020, 40(17): 3803–3807.
丁雅明, 丁亚娜, 孙婉琴, 刘建根. 从中国家族性高胆固醇血症患者的诊断及治疗现状谈慢病管理. 中国老年学杂志, 2020, 40(17): 3803–3807.
[8] Teng HB, Gao Y, Guo YL, Wu CQ, Zhang HB, Zheng X, Lu JP, Li Y, Qiao SB, Liu JM. Detection rate and clinical characteristics of familial hypercholesterolemia among Chinese patients with coronary artery disease., 2021, 36(5): 444–450.
滕浩波, 高岩, 郭远林, 吴超群, 张海波, 郑昕, 路甲鹏, 李艳, 乔树宾, 刘佳敏. 我国成人冠心病患者中家族性高胆固醇血症检出率及治疗现状. 中国循环杂志, 2021, 36(5): 444–450.
[9] Chen PP, Chen X, Zhang SY. Current Status of Familial Hypercholesterolemia in China: A Need for Patient FH Registry Systems., 2019, 10: 280.
[10] Abifadel M, Varret M, Rabes JP, Allard D, Ouguerram K, Devillers M, Cruaud C, Benjannet S, Wickham L, Erlich D, DerréA, Villéger L, Farnier M, Beucler I, Bruckert E, Chambaz J, Chanu B, Lecerf JM, Luc G, Moulin P, Weissenbach J, Prat A, Krempf M, Junien C, Seidah NG, Boileau C. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia., 2003, 34(2): 154–156.
[11] Garcia CK, Wilund K, Arca M, Zuliani G, Fellin R, Maioli M, Calandra S, Bertolini S, Cossu F, Grishin N, Barnes R, Cohen JC, Hobbs HH. Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein., 2001, 292(5520): 1394–1398.
[12] Zhu CG, Li S, Wang ZF, Yin KL, Wu NQ, Guo YL, Gao Y, Li XL, Qing P, Liu G, Dong Q, Zhou Z, Li JJ. Homozygous familiar hypercholesterolemia in China: Case series from the national lipid clinics and literature review., 2017, 14: 75–80.
[13] Atherosclerosis and Coronary Heart Disease Working Group of Chinese Society of Cardiology, Editorial Board of Chinese Journal of Cardiology. Chinese expert consensus on screening, diagnosis and treatment of familial hypercholesterolemia., 2018, 46(2): 99–103.
中华医学会心血管病学分会动脉粥样硬化及冠心病学组, 中华心血管病杂志编辑委员会. 家族性高胆固醇血症筛查与诊治中国专家共识. 中华心血管病杂志, 2018, 46(2): 99–103.
[14] Peng J, Wu X, Wang SL, Zhang S, Wang XM, Liu ZS, Hong J, Ye PC, Lin J. Familial hypercholesterolemia in China half a century: a review of published literature., 2019, 36: 12–18.
[15] Mahdieh N, Heshmatzad K, Rabbani B. A systematic review of LDLR, PCSK9, and APOB variants in Asia., 2020, 305: 50–57.
[16] van der Graaf A, Avis HJ, Kusters DM, Vissers MN, Hutten BA, Defesche JC, Huijgen R, Fouchier SW, Wijburg FA, Kastelein JJP, Wiegman A. Molecular basis of autosomal dominant hypercholesterolemia: assessment in a large cohort of hypercholesterolemic children., 2011, 123(11): 1167–1173.
[17] Cao YX, Wu NQ, Sun D, Liu HH, Jin JL, Li S, Guo YL, Zhu CG, Gao Y, Dong QT, Liu G, Dong Q, Li JJ. Application of expanded genetic analysis in the diagnosis of familial hypercholesterolemia in patients with very early-onset coronary artery disease., 2018, 16(1): 345.
[18] Du R, Fan LL, Lin MJ, He ZJ, Huang H, Chen YQ, Li JJ, Xia K, Zhao SP, Xiang R. Mutation detection in Chinese patients with familial hypercholesterolemia., 2016, 5(1): 2095.
[19] Xiang R, Fan LL, Lin MJ, Li JJ, Shi XY, Jin JY, Liu YX, Chen YQ, Xia K, Zhao SP. The genetic spectrum of familial hypercholesterolemia in the central south region of China., 2017, 258, 84–88.
[20] Teng YN, Pan JP, Chou SC, Tai DY, Lee-Chen GJ. Familial defective apolipoprotein B-100: detection and haplotype analysis of the Arg(3500)→Gln mutation in hyperlipidemic Chinese., 2000, 152(2): 385–390.
[21] Lee C, Cui YX, Song JX, Li SF, Zhang F, Wu MY, Li L, Hu D, Chen H. Effects of familial hypercholesterolemia- associated genes on the phenotype of premature myocardial infarction., 2019, 18(1): 95.
[22] Chiou KR, Charng MJ, Chang HM. Array-based resequencing for mutations causing familial hypercholesterolemia., 2011, 216(2): 383–389.
[23] Yang KC, Su YN, Shew JY, Yang KY, Tseng WK, Wu CC, Lee YT. LDLR and ApoB are major genetic causes of autosomal dominant hypercholesterolemia in a Taiwanese population., 2007, 106(10): 799–807.
[24] Chiou KR, Charng MJ. Common mutations of familial hypercholesterolemia patients in Taiwan: characteristics and implications of migrations from southeast China., 2012, 498(1): 100–106.
[25] Stoekenbroek RM, Kastelein JJP. Proprotein convertase subtilisin/kexin type 9: from genetics to clinical trials., 2018, 33(3): 269–275.
[26] Guo QY, Feng XX, Zhou YJ. PCSK9 variants in familial hypercholesterolemia: a comprehensive synopsis., 2020, 11: 1020.
[27] Shaik NA, Al-Qahtani F, Nasser K, Jamil K, Alrayes NM, Elango R, Awan ZA, Banaganapalli B. Molecular insights into the coding region mutations of low-density lipoprotein receptor adaptor protein 1 (LDLRAP1) linked to familial hypercholesterolemia., 2020, 22(6): e3176.
[28] Tang L. Screening of novel pathogenic genes of familial hypercholesterolemia and effect of soluble epoxide hydrolase gene mutant on the function of LDLR [Dissertation]. HuazhongUniversity of Science and Technology, 2016.
唐玲. 家族性高胆固醇血症患者新致病基因筛查及可溶性环氧化物水解酶基因突变体对细胞LDLR功能的影响[学位论文]. 华中科技大学, 2016.
[29] Harris TR, Hammock BD. Soluble epoxide hydrolase: gene structure, expression and deletion., 2013, 526(2): 61–74.
[30] El-Sherbeni AA, El-Kadi AOS. The role of epoxide hydrolases in health and disease., 2014, 88(11): 2013–2032.
[31] Yang SM, Ke XA, Liang HT, Li R, Zhu HJ. Case report: a clinical and genetic analysis of childhood growth hormone deficiency with familial hypercholesterolemia., 2021, 12: 691490.
[32] Parini P, Angelin B, Lobie PE, Norstedt G, Rudling M. Growth hormone specifically stimulates the expression of low density lipoprotein receptors in human hepatoma cells., 1995, 136(9): 3767–3773.
[33] Khalil YA, Rabès JP, Boileau C, Varret M. APOE gene variants in primary dyslipidemia., 2021, 328: 11–22.
[34] Brown MS, Goldstein JL. Expression of the familial hypercholesterolemia gene in heterozygotes: mechanism for a dominant disorder in man., 1974, 185(4145): 61–63.
[35] Besseling J, Hovingh GK, Huijgen R, Kastelein JJP, Hutten BA. Statins in familial hypercholesterolemia: consequences for coronary artery disease and all-cause mortality., 2016, 68(3): 252–260.
[36] Hamilton-Craig I, Kostner K, Colquhoun D, Woodhouse S. Combination therapy of statin and ezetimibe for the treatment of familial hypercholesterolemia., 2010, 6: 1023–1037.
[37] Qian LJ, Gao Y, Zhang YM, Chu M, Yao J, Xu D. Therapeutic efficacy and safety of PCSK9-monoclonal antibodies on familial hypercholesterolemia and statin- intolerant patients: A meta-analysis of 15 randomized controlled trials., 2017, 7(1): 238.
[38] Santos RD, Gidding SS, Hegele RA, Cuchel MA, Barter PJ, Watts GF, Baum SJ, Catapano AL, Chapman MJ, Defesche JC, Folco E, Freiberger T, Genest J, Hovingh GK, Harada-Shiba M, Humphries SE, Jackson AS, Mata P, Moriarty PM, Raal FJ, Al-Rasadi K, Ray KK, Reiner Z, Sijbrands EJG, Yamashita S, International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel. Defining severe familial hypercholesterolaemia and the implications for clinical management: a consensus statement from the International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel., 2016, 4(10): 850–861.
[39] Gaudet D, Gipe DA, Pordy R, Ahmad Z, Cuchel M, Shah PK, Chyu KY, Sasiela WJ, Chan KC, Brisson D, Khoury E, Banerjee P, Gusarova V, Gromada J, Stahl N, Yancopoulos GD, Hovingh GK. ANGPTL3 inhibition in homozygous familial hypercholesterolemia., 2017, 377(3): 296–297.
[40] Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ, CANTOS Trial Group. Antiinflammatory therapy with canakinumab for atherosclerotic disease., 2017, 377(12): 1119–1131.
[41] Joint committee issued Chinese guideline for the management of dyslipidemia. 2016 Chinese guideline for the management of dyslipidemia in adults., 2016, 31(10): 937–950.
中国成人血脂异常防治指南修订联合委员会. 中国成人血脂异常防治指南(2016年修订版). 中国循环杂志, 2016, 31(10): 937–950.
[42] Thompson GR. Managing homozygous familial hypercholesterolaemia from cradle to grave., 2015, 18: 16–20.
[43] Writing Committee, Lloyd-Jones DM, Morris PB, Ballantyne CM, Birtcher KK, Daly DD, DePalma SM, Minissian MB, Orringer CE, Smith SC. 2016 ACC expert consensus decision pathway on the role of non-statin therapies for LDL-cholesterol lowering in the management of atherosclerotic cardiovascular disease risk: a report of the American college of cardiology task force on clinical expert consensus documents., 2016, 68(1): 92–125.
[44] Zhao L, Wen J, Guo YL. Research progress of liver transplantation for patients with homozygous familial hypercholesterolemia., 2021, 29(4): 353–358.
赵量, 温军, 郭远林. 肝脏移植治疗纯合型家族性高胆固醇血症的研究进展. 中国动脉硬化杂志, 2021, 29(4): 353–358.
[45] Stefanutti C, Julius U, Watts GF, Harada-Shiba M, Cossu M, Schettler VJ, De Silvestro G, Soran H, Van Lennep JR, Pisciotta L, Klör HU, Widhalm K, Moriarty PM, MIGHTY MEDIC Multinational Society. Toward an international consensus-integrating lipoprotein apheresis and new lipid-lowering drugs., 2017, 11(4): 858– 871.e3.
[46] Alim A, Tokat Y, Erdogan Y, Gokkaya Z, Dayangac M, Yuzer Y, Oezcelik A. Liver transplantation for homozygote familial hypercholesterolemia: the only curative treatment., 2016, 20(8): 1060–1064.
[47] Li JJ, Li S, Zhu CG, Wu NQ, Zhang Y, Guo YL, Gao Y, Li XL, Qing P, Cui CJ, Xu RX, Jiang ZW, Sun J, Liu G, Dong Q. Familial hypercholesterolemia phenotype in Chinese patients undergoing coronary angiography., 2017, 37(3): 570–579.
Progress on familial hypercholesterolemia
Wanzi Jiang1, Liwen Zhang2, Caihong He2,3, Meihua Ruan2, Yong Ji4, Jianrong Yu2,3, Hongwen Zhou1
Familial hypercholesterolemia (FH) is an autosomal inherited disease characterized by a significant increase in low density lipoprotein cholesterol (LDL-C), tendon xanthoma and premature coronary artery disease (PCAD). In this paper, we analyze the current research status of FH, summarize the reported mutation gene loci in Chinese FH patients and treatment for them, and elaborate the current status of patents and drug researches. The results show that scientific outcomes of FH are increasing with a good developmental trend and the most popular topics of FH study are pathogenesis, treatment of FH, and research on juvenile FH patients. In terms of patents, large pharmaceutical companies, such as Regeneron Pharmaceuticals Inc, AstraZeneca Plc, Merck & Co Inc, are actively engaged in FH detection, diagnosis and treatment. In addition, 12 drugs have been launched in the United States, Japan, Europe and other countries or regions, bringing hope to FH patients.
familial hypercholesterolemia; rare disease; gene loci; research status
2021-06-22;
2021-08-12
国家重点研发计划项目(编号:2019YFA0802701,2018YFA0506904),国家自然科学基金重大研究计划项目(编号:91854122)和国家自然科学基金专项项目(编号:L1924031)资助[Supported by the National Key Research and Development Program of China (Nos. 2019YFA0802701, 2018YFA0506904), the Major Research Plan of the National Natural Science Foundation of China (No.91854122) and the National Natural Science Foundation of China (No. L1924031)]
蒋琬姿,在读博士研究生,专业方向:肥胖、脂代谢和罕见代谢病。E-mail: 1995jwz@sina.com
张丽雯,硕士,馆员,研究方向:生物情报学。E-mail: zhangliwen@sibs.ac.cn
蒋琬姿和张丽雯并列第一作者。
季勇,博士,教授,研究方向:心血管疾病分子机制及药物防治。E-mail: yongji@njmu.edu.cn
于建荣,硕士,研究员,研究方向:科技情报。E-mail: yujianrong@sibs.ac.cn
周红文,博士,教授,主任医师,研究方向:肥胖、脂代谢和罕见代谢病。E-mail: drhongwenzhou@njmu.edu.cn
10.16288/j.yczz.21-218
2021/9/9 16:52:17
URI: https://kns.cnki.net/kcms/detail/11.1913.R.20210909.1431.001.html
(责任编委: 孟卓贤)
