基于Diels-Alder反应的石墨烯基自修复材料①
2021-11-24阮英波张光喜卢红斌张承双马秀萍霍炳呈
阮英波,张光喜,卢红斌,张承双,马秀萍,吴 凡,霍炳呈
(1.西安航天复合材料研究所,西安 710025;2.复旦大学,上海 200438)
0 引言
固体火箭发动机是航天运载器和导弹武器的主要动力系统,发动机壳体等聚合物基复合材料构件在生产制造的过程中难免会产生缺陷,如果能对这些缺陷进行修复,有望提高成品率和生产效率,降低成本;同时,发动机在储存、运输和服役过程中也会因为各种原因产生复合材料构件损伤,影响构件的承载能力和使用性能,引发安全隐患,如果能对产生的损伤进行有效修复,则可以提高构件使用安全性,提升导弹武器服役性能,延长使用寿命。因此,复合材料的缺陷/损伤修复技术对固体火箭发动机降低成本、提升性能显得尤为重要。聚合物基复合材料主要由增强纤维和树脂基体组成,要想实现复合材料的损伤修复首先要开发具有高效修复性能的树脂基体。
将生物体损伤后可自修复特征引入到合成材料中,赋予材料自修复能力,可消除缺陷和损伤,延长材料使用寿命[1-4]。利用超分子相互作用和动态共价键等可逆相互作用实现损伤自修复的本征型自修复材料,因具有不需要外加愈合剂和可多次可逆自修复的特点而受到广泛关注[1-4]。与超分子相互作用相比,动态共价键由于键能更高、更稳定,成为本征型自修复材料设计与应用的最佳选择[3,5]。基于动态共价键的自修复过程本质上是动态共价键解离后界面两侧的大分子链通过链段运动实现分子链的相对运动,导致物理界面消失并形成新的动态化学键从而实现材料自修复的过程。因此,强的链段运动能力是实现快速高效自修复的关键,目前研究的自修复材料主要集中在弹性体和凝胶材料等链段可快速运动的材料体系中[1-4]。但这类材料的力学强度通常较低,限制了其在很多领域的应用。与弹性体和凝胶材料相比,聚合物塑料具有更优异的力学性能和更弱的链段运动能力,因而如何实现塑料材料的高效自修复依然存在一定挑战。
塑料材料的特点是室温附近聚合物链处于玻璃态或结晶态,无法通过链段运动实现聚合物链的扩散和运动,难以实现材料的自修复。只有当温度超过玻璃化转变温度或熔点后,链段具有了较强的运动能力,材料才能具有自修复性能。因此,塑料材料的自修复必须在高温下进行,这就要求所采用的动态共价键需要有高温下可解离的特点。Diels-Alder反应(D-A反应)是共轭双烯与取代烯烃反应生成取代环己烯的反应,该反应是可逆的,在120 ℃附近会发生逆D-A反应,是比较理想的实现塑料材料自修复的可逆反应[6-7]。但是,该温度下塑料材料聚合物链的运动能力通常较弱,自修复速度和修复程度通常较低。石墨烯由于具有优异的电学[8]、热学[9]、光学[9]、力学[10]以及其他性能[11-12]而被广泛应用于聚合物复合材料领域[13-14]。利用石墨烯优异的光-热、电-热和热传导性能,可以显著提高聚合物材料的自修复性能[15-18]。但是,石墨烯与聚合物基体间没有强的相互作用,难以解决石墨烯在聚合物基体中的分散和多次修复过程中石墨烯的再聚集问题[19]。因此,如果能解决石墨烯在聚合物基体中的稳定分散问题,将D-A反应的高温修复特点和石墨烯的促进聚合物自修复的特点相结合,将有望实现塑料材料的高效快速自修复。此类高效率自修复材料有望应用于固体火箭发动机壳体等复合材料构件中,提升复合材料在成型和使用过程中的抗损伤性能和适应性,实现固体火箭发动机复合材料的智能化。
本文通过向糠基功能化的氧化石墨烯(GO-Fu)/糠基功能化的聚合物(Polymer-Fu)复合体系中加入N,N’-4,4’-二苯甲烷双马来酰亚胺(BMI),利用糠基和马来酰亚胺基团之间的D-A反应,将石墨烯片层通过共价键的方式引入聚合物基体中,解决石墨烯片层在聚合物基体中的分散问题。同时,将D-A反应的可逆修复和石墨烯的促进自修复的特点相结合,制备了石墨烯基自修复塑料材料,并对所制备材料的力学性能、松弛行为和自修复性能进行研究。
1 实验
1.1 实验原料
GO由改性Hummers法[20]制备得到。糠胺(FA)、甲基丙烯酸乙酯(EMA)、甲基丙烯酸丁酯(BMA)、甲基丙烯酸糠酯(FMA)等单体为分析纯,购自TCI,经减压蒸馏除去阻聚剂后使用。偶氮二异丁腈(AIBN)、BMI、N,N’-二甲基甲酰胺(DMF)、二甲苯、无水甲醇、无水乙醇等化学试剂均为分析纯,购自国药集团化学试剂有限公司,AIBN经无水乙醇重结晶并干燥后使用,其他试剂购买后直接使用。
1.2 样品制备
GO-Fu制备:GO-Fu由GO与FA反应制备得到(如图1所示)。一个典型的合成过程如下:将0.4 g GO分散在100 ml去离子水中并水浴超声30 min使其充分剥离和分散形成GO分散液;将上述GO分散液和0.2 g FA加入250 ml三口瓶中,混合均匀后在N2保护下于100 ℃反应24 h;产物经过滤并反复用DMF洗涤除去未反应的FA后得到黑色湿态产物,经60 ℃真空干燥24 h后得到GO-Fu。

图1 GO-Fu合成流程图
含糠基侧基的聚甲基丙烯酸酯共聚物(PMA-Fu)制备:PMA-Fu由EMA、BMA和FMA经溶液聚合得到(如图2所示)。一个典型的聚合过程如下:将15.0 g EMA(0.136 mol)、3.5 g BMA(0.024 mol)、1.4 g FMA(0.008 mol)、90 mg AIBN和40 ml二甲苯加入到100 ml单口瓶中,于80 ℃油浴中反应18 h后冷却降温停止反应。反应物经二甲苯稀释至100 ml,于1000 ml无水甲醇中沉淀3次并干燥得到最终产物。

图2 PMA-Fu合成流程图
自修复材料制备:自修复材料由GO-Fu、PMA-Fu和BMI制备得到(如图3所示)。一个典型的制备过程如下:将5 mg GO-Fu和15 ml DMF加入到20 ml样品瓶中,经搅拌超声分散得到均匀分散液,向上述分散液中加入1 g PMA-Fu和75 mg BMI,经搅拌超声分散得到均匀分散液,该分散液在200 ml去离子水中沉淀并过滤干燥得到物体粉末,经150 ℃热压成型成所需尺寸的样条。

图3 基于Diels-Alder反应的石墨烯基自修复材料合成及自修复示意图
1.3 仪器与表征
X射线衍射(XRD):采用PANalytical公司型号为PANalytial X’Pert PRO的X射线衍射仪测试。采用Cu Ka靶(λ= 0.154 nm),扫描衍射角2θ为2~90°。
傅里叶变换红外光谱(FTIR):采用Nicolet 6700测定。
X射线光电子能谱(XPS):采用RBD upgraded PHI-5000C ESCA system(PerkinElmer)测试,采用 Mg Kα 靶(hν=1253.6 eV)。
热失重分析(TGA):采用TA PE Pyris-1测试,在氮气气氛中测试,流速40 ml/min,升温速率10 ℃/min。
原子力显微镜(AFM):采用Bruker Multimode Nanoscope ⅢA测定,测试在室温下采用tapping模式进行。
流变测试:采用Thermofisher公司的HAKKE MARS III流变仪进行测试,动态温度扫描测试条件:直径25 mm平行板夹具,测试频率10 rad/s,测试温度范围60~150 ℃,升温速率10 ℃/min。
力学性能测试:采用CMT-4102的单轴拉伸装置于室温条件下测试,拉伸速率0.5 mm/min。
光学显微镜(OM):采用Leica DM 2500P光学显微分别在普通模式下测试。
2 结果与讨论
2.1 GO-Fu合成及性能表征
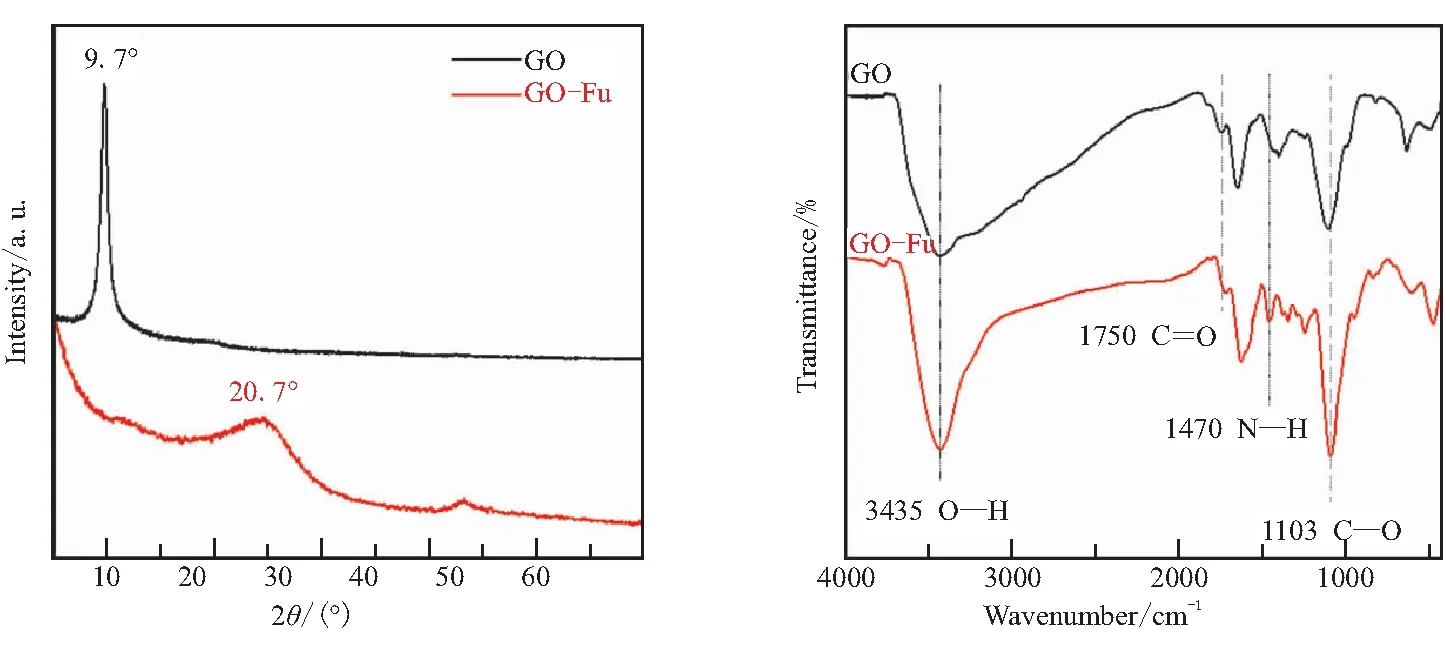
如图1所示, FA通过端氨基与GO表面的环氧基发生开环加成反应接枝到石墨烯片的表面,同时还原GO得到GO-Fu。GO和GO-Fu的XRD、FTIR、XPS及TGA曲线如图4所示,AFM结果如图5所示。
(a)XRD curves (b)FTIR curves

(a)GO (b)GO-Fu
从XRD表征结果(图4(a))可以看出,与GO在9.7°位置存在尖锐衍射峰不同,GO-Fu在10°附近没有衍射峰,相应的衍射峰移动到了低于5°的区域,可见大量糠基接枝物的存在,使得GO的层间距明显增大;同时,GO-Fu在20.7°出现了一个宽的衍射峰,这是还原氧化石墨烯的特征峰,证明接枝过程中GO被还原成了还原氧化石墨烯。
从图4(b)可见,与GO相比,GO-Fu在1470 cm-1处出现了N—H振动峰,这是FA与GO反应过程中引入仲胺基导致的。GO-Fu的XPS谱图(图4(c))可以看到N1s峰,表明GO-Fu中含氮原子。这些结果都表明,成功将FA接枝到了GO表面,从而获得了GO-Fu。
本文通过TGA测试确定 GO-Fu中FA的接枝量。从图4(d)中GO的TGA曲线可以看出,GO 的含氧基团在 350 ℃前已基本失重完全。所以,GO-Fu的TGA曲线 350 ℃以后的重量下降是接枝 FA 的失重。从图中可看出,FA的接枝量大约40%,可见有大量的FA通过共价键接枝到了GO表面。
AFM结果表明,经 FA功能化之后的 GO-Fu 片层的平均厚度由GO的0.8 nm(图5(a))增加到了1.8 nm(图5(b)),这是因为表面大量FA接枝物的存在显著增加了片层的厚度。同时,可以看到GO-Fu的表面是凹凸不平的,这可能是FA在石墨烯片表面的非均匀接枝导致的,这也间接证明 GO 表面的含氧官能团(特别是环氧基)分布是不均匀的。
2.2 PMA-Fu合成及性能表征
以PMA-Fu为例验证复合材料的自修复性能。PMA-Fu合成过程如图2所示,通过EMA、BMA和FMA共聚的方法制备PM-Fu,加入FMA共聚是为了实现在聚合物链上引入糠基的目的(FMA的加入量占单体总量的摩尔百分含量为5%)。通过改变共聚体系中的BMA含量,调控聚合物基体的玻璃化温度和力学性能,如图6所示。

(a)Glass transition temperature(Tg) (b)Stress-strain curves
从图6(a)可以看出,随着共聚体系中BMA含量的增加,所得聚合物的玻璃化温度逐渐降低。当BMA的摩尔百分含量为0时,所得聚合物的玻璃化温度为86 ℃;当BMA的摩尔百分含量增加至15%和30%时,聚合物的玻璃化温度分别降低至67 ℃和54 ℃。与EMA相比,BMA具有更大的柔性侧基,因此BMA的引入,相当于在体系中加入了“增塑剂”,导致玻璃化温度降低[21]。图6(b)表明,BMA摩尔百分含量增加至15%后,聚合物的屈服应力没有明显变化,但断裂伸长率明显增加;但当BMA摩尔百分含量进一步增加至30%时,聚合物的屈服应力降低至14.5 MPa,断裂伸长率进一步增加,达到181%。可见,随着BMA含量增加,聚合物玻璃化温度降低,韧性提高,断裂伸长率显著增加。为了获得具有适中玻璃化温度和优异断裂延伸率的聚合物基体,最终确定加入BMA的摩尔百分含量为15%。
2.3 力学性能与流变行为
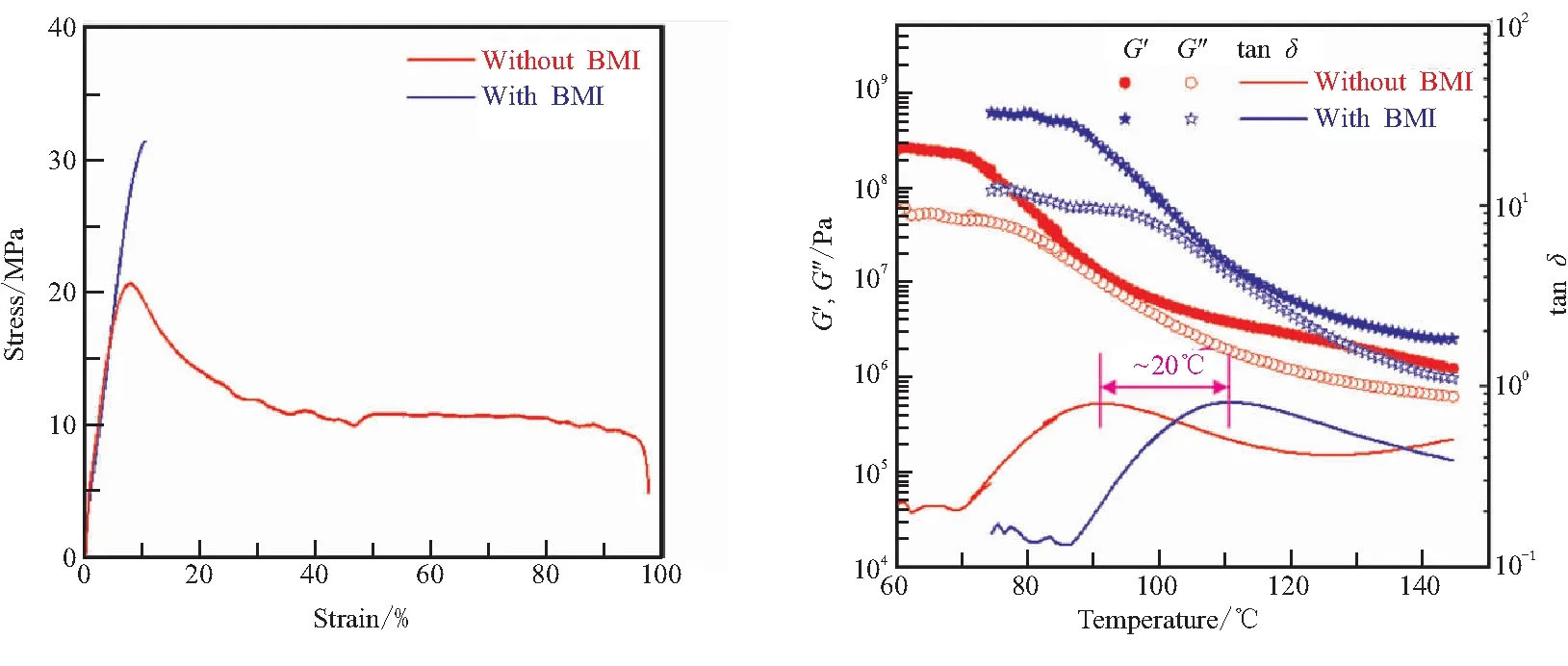
如图3所示,当向PMA-Fu中加入BMI后,PMA-Fu侧基(糠基)中的共轭双键会和BMI的马来酰亚胺基团中的双键发生D-A反应[6-7]。图7(a)对比了BMI的加入对PMA-Fu力学性能的影响。可以看出,随着BMI的加入,聚合物体系的力学强度明显增加,屈服应力由20 MPa增加到32 MPa,提高了60%。与此同时,材料的断裂延伸率由97%降低至10%,材料也由韧性破坏转变为脆性破坏。这是因为BMI的加入起到了交联剂的作用,让原本线性结构的PMA-Fu交联形成了具有三维网络结构的交联体系(见图3),所以力学强度提高而断裂伸长率显著降低。值得注意的是,BMI的加入不仅带来了材料力学性能的转变,也改变了材料的松弛行为。图7(b)是加入BMI前后材料线性流变性能数据。可以看出,加入BMI后,材料的玻璃化温度增加了约20 ℃,同时可以看到,加入BMI后玻璃态下的储能模量(G′)和损耗模量(G″)都有明显提高。这是因为BMI加入导致体系交联密度增加,显著抑制了PMA-Fu链段的运动,带来玻璃化温度和模量的升高。
(a)Stress-strain curves (b)Rheological behavior
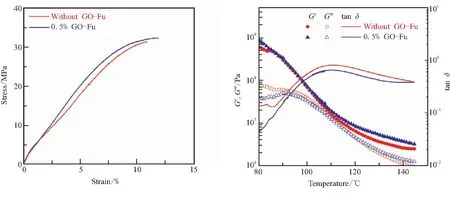
在此基础上,进一步考察了GO-Fu加入对材料力学性能和流变行为的影响。从图8(a)可以看出,向PMA-Fu/BMI体系中加入质量含量0.5% GO-Fu后复合材料的拉伸强度和断裂延伸率略有提高,但没有显著变化。这可以间接证明GO-Fu在聚合物基体中分散良好,没有出现明显团聚,否则会带来力学性能的降低。从线性流变曲线上同样可以看出,加入质量含量0.5% GO-Fu后,复合材料的松弛行为没有明显变化。可见,少量GO-Fu没有明显改变材料的力学性能和松弛行为。
(a)Stress-strain curves (b)Rheological behavior
2.4 自修复性能
为直观展示D-A反应的可逆性,将PMA-Fu、BMI和GO-Fu溶于DMF得到的均匀分散液,考察了分散液的溶胶-凝胶行为。由图9可以看出,分散液在75 ℃下经过10 h反应后,可形成没有流动性的凝胶;如果将该凝胶升温至120 ℃处理1 h,其又可完全转化为具有很好流动性的溶胶;溶胶如果进一步降温至75 ℃/6 h,又可完全转化为凝胶。可见,分散液可以实现溶胶-凝胶的可逆转变。这是因为在75 ℃下,BMI中的马来酰亚胺基团会与PMA-Fu和GO-Fu中的糠基团发生D-A反应,体系会交联形成具有三维网络结构的凝胶;但当温度升高至120 ℃后,体系中又会发生逆D-A反应,重新解离生成PMA-Fu、GO-Fu和BMI,体系又由三维网络结构的凝胶转变为具有很好流动性的溶胶(见图3)。通过该实验可以很好地证明体系的动态可逆过程,这是复合材料实现自修复的基础。

图9 分散体系的溶胶-凝胶转变
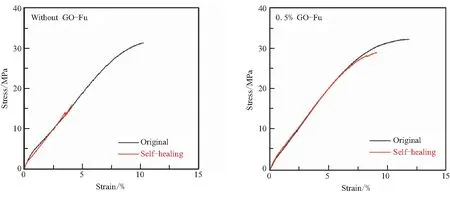
图10分别给出了不加GO-Fu的样品(0% GO-Fu)和加质量含量0.5% GO-Fu的样品(0.5% GO-Fu)的原始试样(original)和破坏后经130 ℃/1 h修复后试样(self-healing)的拉伸应力应变曲线。从图10(a)可以看出,不加GO-Fu试样的原始拉伸强度为31.2 MPa,破坏后经130℃/1 h修复后的拉伸强度仅有15.9 MPa,修复性能较差。与之对应,加入0.5% GO-Fu后,原始拉伸强度和130℃/1 h修复后的拉伸强度分别为32.2 MPa和29.2 MPa,修复后的强度已经比较接近原始强度,表现出很好的自修复性能。
(a)Without GO-Fu (b)With 0.5% GO-Fu
为了定量评价材料的自修复性能,将修复后试样拉伸强度与原始试样拉伸强度的比值定义为修复效率。图11分别给出了不加GO-Fu和加入0.5% GO-Fu试样在130℃修复不同时间后的修复效率。可以看出,材料的修复效率随着修复时间的延长逐渐增加,之后趋于比较恒定的水平,不加GO-Fu材料的修复效率只能达到50%左右,而加入0.5% GO-Fu后材料的修复效率可以接近100%。
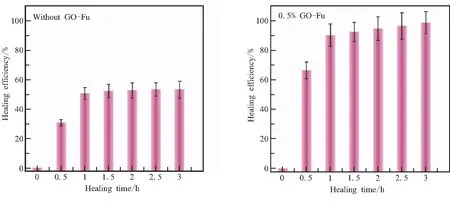
(a)Without GO-Fu (b)With 0.5% GO-Fu
为了直观展示材料的自修复性能,通过显微镜原位观察复合材料损伤表面在130 ℃的修复情况,如图12所示。可以看出,加入GO-Fu样品的原始表面存在大量损伤划痕,随着修复时间的延长,划痕逐渐变模糊直至消失,20 min后,所有划痕都完全消失,材料展现出高效的自修复性能。与之相比,未加GO-Fu样品的修复速度要慢得多,经过20 min后,样品表面仍有大量划痕无法修复。

图12 加入GO-Fu前后复合材料表面划痕随修复时间变化
可以看出,GO-Fu的加入可以显著提高材料的自修复性能。这是因为GO-Fu赋予材料优异的热传导性能,使得外加温度场的能量能够迅速传递到修复界面,促进聚合物链的运动和扩散,提高修复效率[15,18]。同时,每个GO-Fu片层上都含有大量的糠基,可显著增加D-A反应的发生概率,带来修复效率的显著提升。
3 结论
(1)合成了糠基功能化的氧化石墨烯和带糠基侧基的聚甲基丙烯酸酯共聚物,将其与双马来酰亚胺反应,制备了含可逆D-A动态共价键的石墨烯基自修复复合材料。
(2)D-A反应形成的交联网络结构增强了链的刚性,并限制了链段运动,显著提高复合材料的力学性能(强度提高60%)和玻璃化转变温度(提高20℃)。作为交联剂和修复剂的功能化石墨烯的引入,可显著提高复合材料的自修复效率和修复速度,即使功能化石墨烯的加入量仅有0.5%,依然能将材料的修复效率由约50%提高到接近100%。
(3)未来需进一步探索该材料在多种外部刺激(电磁波、激光、电场、红外等)作用下的自修复性能,以丰富自修复材料的应用领域。此外,还需进一步研究如何通过材料结构和化学组成调控实现环氧树脂等热固性聚合物基复合材料的高效自修复。
