过渡金属基催化剂催化5-羟甲基糠醛合成2,5-呋喃二甲醛研究进展
2021-11-18白继峰蒋志薪程曼芳王景芸
白继峰,杨 雨,蒋志薪,程曼芳,张 霖,王景芸*
(1.辽宁石油化工大学 石油化工学院,辽宁 抚顺113001;2.军委后勤保障部军需能源技术服务中心,北京100842)
随着化石能源的日益消耗及环境污染问题的日益严重,可再生能源的开发利用在世界范围内引起广泛重视[1]。生物质是唯一可再生的碳资源,将生物质转化成平台化合物和大宗化学品是生物质转化利用的重要发展方向之一[2]。5-羟甲基糠醛(HMF)是最具价值和潜力的生物基平台化学品,可替代石化工业基础化学品,被视为一种介于生物基化学和石油基化学之间的关键桥梁化合物[3]。HMF含有一个醛基和一个羟基,根据氧化的位置和氧化程度,可以分别氧化为5-羟甲基-2-呋喃甲酸(HMFCA)、2,5-呋喃二甲醛(DFF)、5-甲酰基-2-呋喃甲酸(FFCA)、2,5-呋喃二甲酸(FDCA)[4];当氧化程度更高时,发生C—C断裂,可生成马来酸酐(MA)等[5]。其中,DFF可以应用于合成含尿素的生物基聚合物树脂、含伯胺的聚合物席夫碱、含胺单体、药物化合物、抗真菌化合物、大环分子、荧光材料的底物,因此,被大量应用在医药[6-8]、高分子材料[9-11]、杀虫剂[12]等领域。目前,HMF氧化制备DFF的传统催化体系包括生物酶催化体系、贵金属催化体系和过渡金属催化体系。生物酶催化体系具有反应条件温和、选择性高的特点,但生物酶催化剂在反应介质中往往不稳定,目前可用于工业化应用的生物酶催化剂还太少,且生物酶催化剂开发的周期较长[13];贵金属催化体系催化效率较高,但贵金属价格昂贵,不适用于大规模工业化生产[14-15]。近年来,过渡金属基催化剂被尝试用于HMF氧化制备高附加值呋喃类化合物的反应,并表现出较高的催化活性。相对于贵金属而言,过渡金属价格低廉,资源丰富,具有良好的催化活性和稳定性,是一种极具潜力的催化剂。目前,国内外均有关于HMF氧化制备DFF研究的综述报道,但这些综述有的是对HMF氧化反应研究现状的总体介绍[16],有些是对生物酶和贵金属催化体系等总体综述[17],而对基于过渡金属基催化剂催化的HMF氧化反应研究的综述较少。因此,本文对过渡金属基催化剂催化的HMF氧化制备DFF的反应进行综述和分析。根据催化剂和进行反应所采用的辅助手段进行了详细的分类,分别介绍了锰基、钒基、铜基、铁/钴基催化剂及其他催化体系,从反应条件、反应机理等方面介绍了国内外过渡金属基催化剂催化HMF氧化制备DFF的研究进展。
1 锰基催化体系
锰基催化体系主要有金属盐/溴化物和金属氧化物催化体系,所用氧化剂主要为O2、空气和叔丁基过氧化氢。在反应过程中,催化剂与氧化剂相互作用使氧化剂活化,形成活性过渡态氧,从而增强催化氧化反应的效果。
1.1 金属/溴化物催化体系
HMF具有羟基和醛基结构单元,由于羟基上的氧具有2对孤对电子,容易给出电子,而醛基的碳的电子密度高,容易失去电子,二者均易被氧化。用金属/溴化物催化剂通过空气将羟基氧化成醛基是最为高效的方法。
Partenheimer等[18]在前人利用Co/Mn/Br催化对二甲苯氧化制备对苯二甲酸(PTA)的基础上,研究了醋酸为溶剂,Co/Mn/Br/Zr对催化氧化HMF制备2,5-呋喃二甲醛(DFF)的影响。在特定条件下,DFF的收率可达57%。相对于其他催化体系而言,该催化体系也有一些缺点,例如反应条件苛刻,且在反应过程中HMF易被过度氧化成其他副产物。在选定的条件下,只能得到57%的DFF收率。由于催化效率较低,易产生溴污染,金属盐催化剂的分离困难和DFF的纯化困难,因此使用较少。
1.2 金属氧化物催化体系
1.2.1单金属氧化物催化单金属氧化物分子结构中Mn—O的强度往往不同于其他化合物,能够通过电子转移而使反应物活化。由于Mn2+离子的d电子层容易失去或者得到电子,具有较强的氧化还原性能。因此,锰基金属氧化物被广泛用作各种氧化反应的催化剂。
Nie等[19]报道了在分子氧存在下,在N,N-二甲基甲酰胺(DMF)中用氧化锰和氧化锰八面体分子筛(OMS-2)将HMF氧化为DFF。研究表明:相对于其他锰氧化物催化剂,OMS-2得到了较高的HMF转化率(100%)和DFF收率(97%)。通过比较OMS-2与水钠锰矿型(Na-OL-1)、非晶态二氧化锰(γ-MnO2),以及其他形态的氧化锰分子筛(OMS-7、OMS-6和OMS-1)的活性,发现OMS-2对DFF的选择性最高,并具有良好的活性;尽管Na-OL-1表现出最高的活性,但选择性低于OMS-2。
氧化锰和OMS-2催化HMF氧化反应的机理如图1所示,由图可知,HMF被OMS-2中的晶格氧氧化形成DFF。Mn4+还原为Mn3+的同时,Mn3+又被分子氧再氧化成Mn4+,从而完成了催化循环。通过分析未参与反应和参与反应的OMS-2的X射线光电子能谱(XPS),结果表明:晶格氧质量分数从72.8%降低至65.0%;同时,氧空位从21.4%增加到27.8%,证实了Mn4+/Mn3+的氧化还原循环。
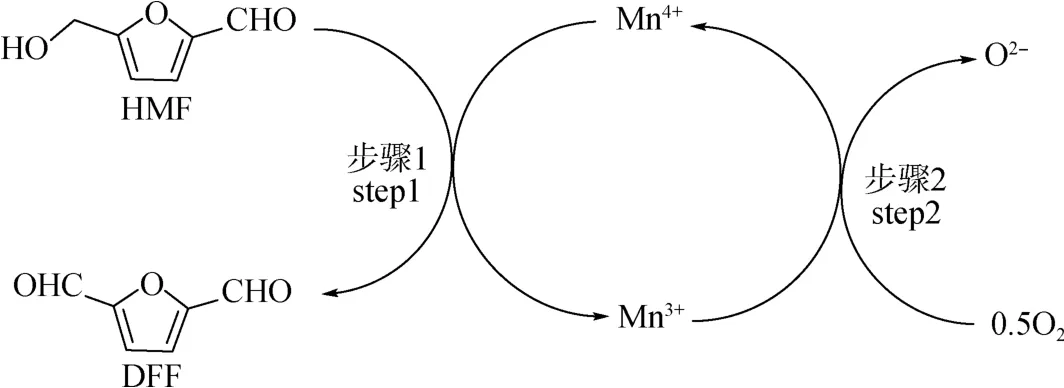
图1 锰氧化物反应的机理[19]Fig.1 Proposed reaction mechanism for the manganese redox cycle[19]
将催化剂负载到具有合适孔结构和比表面积的载体上,不仅可以增强催化剂的机械性能和耐热、传热性,而且可以提高单位质量活性组分的催化效率。同时节省催化剂活性金属的用量,从而降低成本。
相对于普通催化剂,纳米催化剂具有诸多特殊的优势。Liu等[20]在DMF中以Mn3O4·纳米粒子(NPs)为催化剂,以氧气为氧源催化HMF转化为DFF。研究结果表明:Mn3O4·NPs使HMF达到定量转化并且得到83.2%的DFF。与普通催化剂相比,该催化剂因尺寸小、表面所占的体积百分比大、表面的价态和电子价态与反应物内部不同、表面原子配位不全等导致表面的活性位置增加,从而能够更大程度地分散于HMF表面,致使表面光滑程度变差,形成了凸凹不平的原子台阶,增加了化学反应的接触面,这大大增加了催化活性,使HMF的氧化反应更彻底。此外,研究还发现:在相同的反应条件下,当使用分子氧作为氧化剂时,DFF的收率达到82.1%;而叔丁基过氧化氢为氧化剂时,DFF的收率仅为7.2%,原因是叔丁基过氧化氢的强氧化能力导致呋喃环开环。当氧化剂改为双氧水时,产物的产率依然很低,仅为5.3%,原因可能是因为MnO2、Mn5O8和Mn3O4等可以催化过氧化氢的分解。
Biswas等[21]报道了当空气作为氧化剂时,将介孔锰负载于氧化钴上在N,N-二甲基甲酰胺(DMF)中催化HMF转化为DFF。研究结果表明:在130℃的DMF中,分别单独使用介孔MnOx和CoOx作为催化剂时,虽然获得了接近定量的DFF选择性,但是DFF的收率分别仅为13%和18%。在相同的反应条件下,5%(质量分数)的Mn/CoOx使HMF转化率达到了80%,并且DFF选择性达到了96%,反应机理见图2。

图2 HMF氧化成DFF的合理自由基机理[21]Fig.2 Plausible radical free mechanism for the oxidation of HMF to DFF[21]
由图2可见,通过向反应中加入自由基清除剂(2,6-二叔丁基-4-甲基苯酚,BHT)证实该反应通过自由基反应途径进行。CoOx表面上带有Mn,研究人员提出,反应始于HMF的O—H解离;同时,晶格氧质子化。在下一步反应中,从α-CH2中除去质子以产生DFF,金属氧化物随后被还原,产生H2O2,然后H2O2分解为水和氧气。最后,发生还原的金属物种再氧化以完成催化循环。
Ke等[22]使用掺氮的氧化锰(N-MnO2)为催化剂,以分子氧为氧化剂,室温下在甲苯中催化HMF氧化制备DFF,反应6 h后DFF的选择性接近100%。通过扩展X射线吸收精细结构(EXAFs)分析表明:MnO2掺杂氮后Mn—O键略有拉长,降低了Mn—O配位数。N-MnO2催化剂结构的这些变化通过产生更多的表面缺陷位点和协调的不饱和锰位点,显著提高了其将HMF氧化为DFF的催化活性。N-MnO2催化氧化HMF的反应机理如图3所示,反应由表面空位(1)上的O2活化引发,然后HMF通过吸附在配位不饱和Mn位点(2)的OH基团在N-MnO2上解离,并且来自—OH基团的氢物种转移到氮掺杂剂上(3)。来自HMF的—CH2基团的第二个氢在空位(4)处转移到超氧上。最后,DFF从N-MnO2中解吸(5),催化循环在O2分子的帮助下完成(6)。氮掺杂引起的配位不饱和锰位点和表面缺陷在氧化过程中起着重要作用。

图3 N-MnO2催化氧化HMF的机理[22]Fig.3 Proposed mechanism for the oxidation of HMF over the N-MnO2[22]
1.2.2复合金属催化金属的特性会因加入别的金属形成合金而改变,其对化学吸附的强度、催化活性和选择性等效应都会改变。
Neaţu等[23]报道了利用Mn0.7Cu0.05Al0.25组成的Mn和层状双金属氢氧化物铜(CuLDHs)作为催化剂,以分子氧为氧原子供体,在水中催化HMF氧化成DFF。结果发现:在90℃的水中,0.8 MPa O2的条件下,HMF的转化率达到90%,并且获得了78%的DFF;将水换成其他溶剂(例如甲醇、乙腈、N,N-二甲基甲酰胺、甲基异丁基酮和甲苯)后,DFF的收率达到40%~70%。在这项研究中,虽然DFF的产率达到了78%,但是获得了5%的副产物FDCA,以及16%的副产物FFCA,Mn0.70Cu0.05Al0.25可以重复使用5次。Mn0.70Cu0.05Al0.25催化氧化HMF的机理如图4所示,HMF氧化反应由Cu2+还原成Cu+开始,同时释放电子(步骤1)。结果表明:Mn3+到Mn4+的氧化与释放电子是协同作用(步骤2)。此外,随着Mn4+向Mn3+的还原,HMF氧化为DFF。

图4 Mn0.70 Cu0.05 Al0.25催化氧化HMF的机理[23]Fig.4 Proposed mechanism for the oxidation of HMF over the Mn0.70Cu0.05 Al0.25 catalyst[23]
2 钒基催化剂
2.1 负载型催化剂
2.1.1分子筛SBA-15 SBA-15介孔分子筛具有比表面积较高、孔道结构规则及孔径易于调节、热稳定性和水热稳定性较好、表面易改性等优点,在催化化学和吸附分离等领域有很大的应用前景。
Liu等[24]利用固定在SBA-15上的氨基官能化的Cu2+(SBA-NH2-Cu2+)和VO2+(SBA-NH2-VO2+)作为催化剂,在110℃的甲苯和4-氯甲苯中,以分子氧为氧源,SBA-NH2-VO2+和SBA-NH2-Cu2+分别获得了36%和9%的DFF。在相同的反应条件下,物理混合的SBA-NH2-VO2+和SBA-NH2-Cu2+获得了大约62%的DFF,副产物FDCA和HMFCA的产率为27%和4%。由于催化剂负载在SBA-15上,不会造成反应后催化剂与反应体系难分离的问题。
2.1.2石墨氮化碳(g-C3N4) 近年来,光催化技术的研究在逐渐从可再生清洁能源领域扩展到传统催化化学的各领域中[25-26],光催化剂的种类也变得越来越多。其中石墨氮化碳(g-C3N4)具有独特的电子结构和优异的化学稳定性,近年来常被用作不含金属组分的催化剂和催化剂载体。在能源和材料领域,g-C3N4逐渐引起人们的关注[27]。
Chen等[28]以V-g-C3N4为催化剂,氧气为氧源,在DMF中催化HMF的氧化反应。结果发现:6 h后HMF达到定量转化且DFF的产率达到82%,并用其他金属(例如Mo、Fe、Ni、Co、Mn和Cu)替换V,DFF的产率降低至3%~23%。通过XPS分析检测,证明了发生VV-VIV氧化还原循环,并且与Ding等[29]的研究结果一致。
2.2 含钒的磷酸氧化物
磷酸催化剂的催化产率较低,且反应时间长,为达到提高特定目标产物产率的目的,许多研究者采用了磷酸-氧化物或磷酸-有机铵离子催化氧化HMF,并取得了不错的效果。
Grasset等[30]报道了以分子氧作为氧化剂,在110℃的甲苯中使用嵌入到磷酸钒氧化物(C14VOPO4·2H2O)中的烷基三甲基铵离子催化氧化HMF,可以获得83%的DFF。通过研究发现随着烷基的链长从C10增加到C16,DFF选择性和HMF转化率从85%和76%分别提高到90%和84%。此外,Grasset等还研究了溶剂对催化活性的影响,研究发现相对于在80~106℃下含氧溶剂(如乙酸叔丁酯、2-甲基四氢呋喃和环戊基甲基醚)得到3%~14%的DFF而言,芳香族溶剂(如二甲苯、氯苯和甲苯)中C14VOPO4表现出更好的活性,在110℃时HMF达到定量转化并且DFF的收率达到63%~82%。
研究人员通过Mars-VanKrevelen机理证明了钒物种之间存在氧化还原循环。与Hanson等[31]的研究结果一致,可能是由于HMF将V5+还原成V4+,以及O2将V4+氧化成V5+。Grasset等[30]通过在甲苯中添加(BHT)进一步证明了该反应是按照自由基机理进行的,发现C14VOPO4活性比不存在自由基清除剂的体系好,在HMF的转化率达到90%的同时,产物也有较高的选择性(>90%)。此外,由于在没有HMF作为反应物的情况下,没有观察到甲苯的氧化产物(苄醇、苯甲醛和苯甲酸),研究人员认为自由基物种是HMF生成副产物的原因,如甲酸、乙酸和5-甲酰氧基甲基糠醛(FMF)。这表明,所形成的基团是从HMF的—CH—OH部分中夺取质子,并由此产生DFF,同时V5+还原为V4+(图5和图6)。

图5 HMF与C14 VOPO4可能的相互作用[32]Fig.5 Possible interaction of HMF with C14 VOPO4[32]
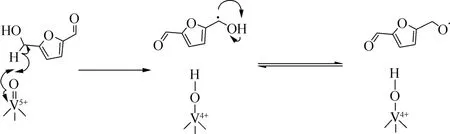
图6 C14VOPO4上DFF可能形成的机理途径[32]Fig.6 Possible mechanistic pathway for the formation of DFF on C14 VOPO4[32]
Lai等[32]制备了不同V/P物质的量比的磷酸钒氧化物(VPO)多相催化剂,以空气为氧化剂,在液相中催化5-HMF选择性氧化制DFF,在最优的条件下,DFF产率为83.6%,HMF转化率高达100%。此外经过研究发现:溶剂在HMF的转化和DFF的选择性中起着关键作用。VPO作为异质催化剂,为可持续制备有价值的生物质平台化合物开辟了一种新途径。
2.3 复合金属催化剂
Zhao等[33]通过水热法合成了含有二氧化钒(VO2)的介孔炭球(V-CS),以分子氧为氧化剂,在DMSO中催化HMF氧化制备DFF,在最优的条件下,HMF转化率和DFF产率分别达到100%和99%。采用场发射扫描电镜(FESEM)、BET、XRD、FT-IR、拉曼光谱、X射线吸收近边结构(XANES)和元素分析等方法研究了催化剂的理化性质,通过研究发现催化剂的高活性归因于其比表面积大的介孔结构,使反应物和产物能够自由运动,增强了反应物与活性位点之间的接触。通过热处理,作为活性催化中心的二氧化钒物种高度分散在介孔炭基质中,提高了催化活性。此外,催化剂在回收试验中活性没有明显损失,显示出良好的可用性。
钒基催化剂表现出较好的催化活性的原因是由于V具有不同的价态,从而具有丰富的配位方式。所以钒基催化剂的结构和反应变得复杂[34]。
3 铜基催化剂
除了锰基、钒基催化剂外,铜基催化剂也被应用于HMF的氧化反应中。铜基催化剂催化HMF氧化制备DFF的反应主要包括两类:均相催化体系,如VOSO4和Cu(NO3)2在乙腈中组成的催化体系;非均相催化体系,如纤维素、分子筛等负载型催化剂。
3.1 均相催化剂
Jia等[35]以Cu(NO3)2和VOSO4共同组成的催化体系,以乙腈为溶剂,在80℃、0.1 MPa O2条件下,催化HMF制备DFF。研究发现:在相同的催化剂体系下,加入其他助催化剂后,例如Ce(NO3)3、Ni(NO3)2、Mn(NO3)3、Co(NO3)2、NaNO3和Fe(NO3)3替代Cu(NO3)2,DFF的选择性降低至11%~75%,HMF转化率为11%~34%。在相同的条件下,将乙腈改为甲苯、四氢呋喃、二氯甲烷、N,N-二甲基甲酰胺、二甲基亚砜、异丙醇、甲醇、乙醇和水后,Cu(NO3)2和VOSO4催化体系的催化活性并未提高。同时,提出Cu(NO3)2在VOSO4催化氧化HMF产生DFF的过程中起着至关重要的作用,从而促进Cu(NO3)2降解形成NOx,这有助于在反应过程中将V4+氧化为V5+活性物质(图7)。最初,Cu(NO3)2分解成NOx,然后,VOSO4被氧化生成活性物质V5+,从而将HMF氧化成DFF。然后,由Cu(NO3)2分解形成的Cu2+离子可以有效地防止HMF的C—C裂解,并避免不需要的自由基反应以保持DFF选择性。通过UV-Vis分析证实了这一点,在相同条件下,用其他金属硝酸盐替代Cu(NO3)2后,未发现其他金属硝酸盐降解产生NOx。

图7 VOSO4催化的Cu(NO3)2氧化HMF的反应机理[35]Fig.7 Proposed reaction mechanism for HMF oxidation with Cu(NO3)2 catalyzed by VOSO4[35]
由于N-羟基邻苯二甲酰亚胺(NHPI)含有N—OH,容易生成自由基,常被用作液相催化氧化反应的催化剂。NHPI作为催化剂具有反应条件温和、转化率和选择性高以及对环境友好等特点,具有广阔的应用前景。
Kompanets等[36]研究了NHPI作为助催化剂,在50℃、0.1 MPa O2条件下,在乙腈中Cu(NO3)2催化HMF制备DFF,DFF产率达到了71%。研究人员提出,NHPI首先通过形成相应的邻苯二甲酰亚胺-N-氧基(PINO)自由基将Cu2+还原为Cu+,然后PINO从HMF的羟基部分提取质子,以促进DFF的形成。在相同的反应条件下,当助催化剂同为NHPI时,其他金属催化剂(例如钴基、锰基和钒基的乙酰丙酮配合物)与Cu(NO3)2相比,DFF的产率约为4%~26%。因此,与其他金属催化剂体系相比,铜基催化剂与助催化剂NHPI之间起着协同促进的作用。
Tong等[37]研究了碘酸铜盐在介质(如NHPI、TEMPO(2,2,6,6-四甲基哌啶-1-氧自由基)、1-羟基苯并三唑(HBT)和N-甲基吗啉N-氧化物)存在下催化HMF选择性氧化。在所使用的介质中,HBT与碘酸铜结合后,在130℃DMSO中,0.3 MPa O2的条件下,10 h后HMF的转化率为93%,DFF的选择性为99%。在保持选择性的同时,用CuBr和CuCl代替碘酸铜时,HMF的转化率下降到88%~91%。此外,据Tong等[37]报道,相对于H2O2和t-BuOOH而言,分子氧提高了DFF的收率。碘酸铜-HBT催化HMF制备DFF反应机理如图8所示。
Jia等[38]报道了以Fe(NO3)3/Cu(NO3)2作为催化剂、空气作为氧化剂和K2S2O8作为促进剂将HMF氧化为DFF的有效方法,在80℃的乙腈中,3 h后DFF的产率达到了99%。反应机理如图9所示。
由图9可知,最初,HMF中羟基的氧原子与Fe(III)离子吸附在Fe(NO3)3的表面,通过释放H+得到中间体1。随后,HMF中的α-H被NO3-中的氧原子提取形成中间体2,伴随着电子转移。最终,中间体2经过β-H消除和还原消除产生DFF和Fe2+,伴随着NO2的释放。生成的Fe(II)被空气中的O2和释放的NO2重新氧化为Fe3+。在此过程中,Fe2+和Fe3+的氧化还原循环通过Cu2+和Cu+的氧化还原循环在氧气辅助下的协同作用得到促进。

图9 Fe(NO3)3/Cu(NO3)2催化HMF氧化制DFF的反应机理[38]Fig.9 Plausible mechanism for oxidation of HMF to DFF with Fe(NO3)3/Cu(NO3)2 as catalysts[38]
3.2 非均相催化剂
均相催化体系主要是在有机溶剂中,利用其溶解性,使催化剂与反应物达到分子层面的接触,提高了选择性。但是产品与反应体系不易分离,催化剂不易重复使用,且易产生环境污染物。将活性组分负载在适当的载体上的非均相催化剂可以解决上述问题。
纤维素多孔载体是以纤维素为基质的材料,其亲水性和亲生物性较好,而且通过适当成型方法得到的内部结构有许多网状的相互连通的小孔通向载体表面。Baruah等[39]将铜纳米颗粒与Bi(NO3)3·5H2O一起负载在纤维素上作为催化剂,对HMF生产DFF有显著的催化性能,在80℃乙腈中,在空气作为氧化剂存在下,DFF产率为82%,HMF转化率为85%。该催化剂体系的优点是HMF不会氧化成其他氧化产物,例如HMFCA、FFCA和FDCA。
经过研究发现,金属离子引入分子筛体系,能提高分子筛的反应性能,为进一步的反应提供条件,此外,金属离子在体系中还会产生一定的氧化还原性,对反应有促进的作用。已经报道一系列含铜丝光沸石(MOR)用作将HMF转化为DFF的催化剂载体[40]。在充满分子氧的密闭空间中,于120℃的DMSO中测试了将铜结合到MOR框架中(Cu-MOR),与MOR交换的铜(Cu(Ex)-MOR)以及浸渍在MOR(Cu(im)-MOR)的催化活性。HMF转化率为69%~98%,DFF产率为2%~34%。在相同的反应条件下,在不使用铜的情况下,MOR上的V2O5(质量分数10%)在HMF完全转化时提供63%的DFF,负载在Cu-MOR上的V2O5(10%)能得到92%的DFF,并具HMF达到了定量转化。因此,根据Mars-vanKrevelen机制,表明钒和铜存在协同作用。
4 铁、钴基催化剂
虽然相对于锰基、钒基和铜基催化剂而言,钴、铁基催化剂的用量相对较少。但是近年来,钴、铁过渡金属及其化合物被广泛应用于HMF的氧化反应中,并表现出较好的催化效果。在HMF催化制备DFF的反应中,常用铁、钴的配合物和磁性氧化物作催化剂。
4.1 铁、钴的配合物催化剂
铁、钴等过渡金属配合物对氧化反应具有良好的催化作用,尤其是钴的金属卟啉配合物对HMF氧化反应具有一定的催化作用,但配合物催化属于均相催化,催化剂与反应体系分离困难。因此,可以将金属配合物负载化,形成固载化的金属配合物催化剂。
Gao等[41]将钴卟啉配合物负载在Merrifield树脂上得到催化剂Merrifield-Co-Py,将该催化剂用于催化HMF氧化反应,分析了不同氧化剂、不同溶剂对反应的影响,结果表明:叔丁基过氧化氢和乙腈是最适合的反应氧化剂和溶剂,在最优化条件下,可得到43%的DFF收率,催化循环使用6次,活性无显著降低。
Amarasekara等[42]制备了一种Mn-salen席夫碱,并将其用作催化HMF制备DFF氧化反应的催化剂。室温下,在磷酸盐和CH2Cl2组成的双向溶剂中催化HMF制备DFF。优化条件下,DFF产率达到了89%。但是该催化体系由于使用了NaOCl作为氧化剂,产生了有毒有害的废弃物。
金属卟啉配合物对HMF氧化反应表现出良好的催化活性和选择性,并且固载化解决了催化剂不易循环使用的问题。但此类催化剂配体和配合物的制备过程复杂,反应条件苛刻,合成成本较高,其在HMF氧化反应中的应用受到限制。
4.2 磁性氧化物催化剂
铁、钴等磁性氧化物催化剂制备过程简单,合成成本较低,同时,可以通过外部施加磁场使催化剂与反应混合物快速分离。因此,近年来铁、钴磁性氧化物催化剂受到了研究者的广泛关注。
Ranganath等[43]将炭黑沉积在磁铁矿上制备了(Fe3O4@C)催化剂,在温和的条件下催化HMF氧化制备DFF,在最优条件下,DFF的选择性达到95%。该催化剂在制备过程中仅仅使用了过渡金属,不仅可以将HMF选择性氧化为DFF,还可以对葡萄糖/果糖的转化有一定的催化活性。
Karimi等[44]使用可磁分离的2,2,6,6-四甲基哌啶氧化物(TEMPO)氧化HMF。研究人员通过4-羟基-TEMPO与γ-氨基丙基官能化的Fe3O4@SiO2-NH2的反应,将TEMPO化学接枝到磁性载体上。使用Fe3O4@SiO2-TEMPO催化剂(2%TEMPO)、亚硝酸叔丁酯(5%)作为助催化剂,乙酸(25%)作为添加剂,在50℃的甲苯中,0.1 MPa O2的条件下,反应18 h后DFF的选择性高达99%。
Fe3O4@SiO2-TEMPO催化HMF氧化制备DFF的反应机理如图10所示,该反应需要结合两个氧化还原循环。首先,TEMPO通过氢键与Fe3O4@SiO2-NH2结合并形成电子供体复合物。然后,亚硝酸叔丁酯在乙酸的存在下形成亚硝酸,亚硝酸又分解成NO2和H+。供体复合物被H+夺取电子的同时形成了具有稳定空穴的正电荷结构。具有正电荷结构的电子供体复合物将HMF氧化成DFF,同时被还原成氧铵阳离子,其在NO2还原成NO的过程中被氧化成稳定空穴的正电荷结构的复合物。同时NO又被O2氧化成NO2。就这样,在两个氧化循环反应的结合中,催化反应得以循环进行。此外,该催化剂可以通过外加磁场回收和重复使用,连续重复使用5次后催化性能无明显改变。

图10 使用Fe3 O4@SiO2-TEMPO催化剂将HMF氧化为DFF的反应途径[44]Fig.10 Proposed reaction pathway for the aerobic oxidation of HMF into DFF with Fe3 O4@SiO2-TEMPO as catalyst[44]
Fang等[45]将从金属-有机骨架(MOF)衍生的碳载体上含有Fe和Co的纳米复合材料为催化剂,在100℃甲苯中,1 MPa O2条件下,以Na2CO3作为碱,催化HMF制备DFF。研究发现:在特定的条件下,用FeCo/C(500)将HMF转化为定量收率的DFF。在相同的反应条件下,使用在不同温度下处理得到的FeCo/C(600)、FeCo/C(700)和FeCo/C(800)为催化剂,DFF的收率都很低。通过观察这4种催化剂的TEM图像,Fang等[45]提出FeCo/C(500)是空心NP的针状,这可能是其具有高催化活性的原因。但是随着处理温度的升高,产率反而下降的原因是因为中空结构塌陷并发生了NP的团聚。
5 其他催化体系
除了上述提及的Mn、V、Cu、Co、Fe等催化剂以外,Ce、Mo基金属催化剂在HMF氧化反应中的应用也有报道。Mo在催化领域的应用研究已有相当长的历史,含有Mo的催化剂早已在工业上发挥重要作用。相比于膦-金属配合物,N-杂环卡宾金属配合物对水和空气稳定,由于卡宾碳-金属的键能大,在加热的情况下也不易解离(除N-杂环卡宾的给电子能力更强,易于提高中心金属的电子密度外);N-杂环卡宾的取代基容易调节,便于设计不同的空间大小。
Zhao等[46]用Mo-HNC金属配合物催化HMF制备DFF,在140℃的DMSO中,以氧气为氧源,反应12 h后13%的Mo-HNC使HMF转化率达到97%,并且获得了96%的DFF。随着Mo-HNC中钼的含量减少,DFF的收率也降低。当Mo-NHC中Mo为3%时,DFF收率仅为64%。在另一项研究中,Liu等[47]将铯交换的钼和含钒的杂多酸(CsH4PMo10V2O40)在120℃的DMSO中,0.1 MPa O2条件下催化HMF氧化反应,6 h后DFF的收率达到了定量。研究人员推测,VOx>MoOx可能有利于引起更大的电子离域,以促进HMF氧化。
含钒杂头酸(PMoVHPAs)是高活性氧化催化剂,其活性可通过改变五价钒(VV)的含量来调节。Rodikova等[48]制备了多种具有不同VV量和外球阳离子类型的PMoV HPAs,并作为催化HMF氧化制备DFF的催化剂。以空气为氧化剂,在110℃的水/MIBK混合溶剂中,90 min后DFF的产率为92%。研究发现:PMoV HPAs对HMF氧化的催化性能与其酸度和VV含量有关。该催化剂至少可回收和重复使用5次,进一步研究还发现:外球阳离子的类型和数量可以提高DFF的产率。
吕宏缨等[49]报道了一种HMF氧化制备DFF的方法,具体方法为:将0.050 4 g HMF加入7 mL芳香烃类溶剂溶液中,搅拌后再加入40~120 mg的Anderson型杂多酸盐CeCu(OH)6Mo6O18催化剂,紧接着通入氧气,在110~150℃下进行HMF氧化反应,得到含有DFF的混合液体,然后将混合液进行稀释并使用高效液相色谱仪对产物进行分析。结果表明:该工艺过程不仅可以高效地合成DFF,而且反应条件温和,催化剂可以循环使用,是一种廉价且高效的DFF制备方法。此外,该方法所用的催化剂相比现有技术中的催化剂,DFF选择性更高,而且更加经济。
以上催化体系均需要在碱性水溶液中进行,需要大量酸中和反应液,易产生工业废水。Gupta等[50]报道了一种碳基材料(CSTi)是在水相中将HMF氧化成DFF的极佳催化剂。以氧气为氧源,在70℃的水中反应8 h后HMF转化率为91%,同时获得了88%的DFF。相对于其他催化体系而言,该催化体系反应时间短、反应条件温和、对环境无污染,但是由于使用纯氧为氧化剂,反应不易操作。
6 结 语
HMF可通过生物质碳水化合物脱水得到,其分子中含有醛基和羟基,通过氧化反应可得到多种高附加值精细化学品,而对HMF羟基的选择性催化氧化可制备平台化合物DFF。Mn、V、Cu、Fe和Co等过渡金属基催化剂对HMF氧化制备DFF的反应具有良好的催化活性、选择性和稳定性,可替代或部分替代贵金属完成HMF向DFF的选择性氧化转化,但催化过程仍存在一些问题。如金属盐催化剂不易回收重复使用,钒基催化剂不环保,负载型金属配合物、金属-有机骨架等新型固体催化剂表现出良好的催化性能,但制备过程复杂、合成成本较高;多数HMF氧化反应须在有机溶剂中进行,有机溶剂使用使反应经济性下降,且存在环境污染隐患;HMF氧化反应氧化剂多为氧气、空气、有机过氧化物和过氧化氢,以氧气和空气为氧化剂时,所需气体压力较大,操作安全性较差。因此,设计合成绿色高效的非贵金属基催化剂,在中性水溶液中,以双氧水或空气为绿色氧化剂,在温和条件下进行HMF催化氧化反应,实现HMF向DFF的高选择性转化,是未来研究的发展方向。
