气相色谱-质谱法测定药用丁基橡胶塞中18种多环芳烃的含量
2021-11-15薄晓文郭利娟刘雪婷
薄晓文, 张 敏, 郭利娟, 刘雪婷, 沈 永*
(1.山东省医疗器械和药品包装检验研究院,山东济南 250101;2.国家药品监督管理局药品包装材料质量控制重点实验室,山东济南 250101;3.山东省医疗器械生物学评价重点实验室,山东济南 250101)
多环芳烃(Polycyclic Aromatic Hydrocarbons,PAHs)具有致癌、致畸和致突变等危害,并且具有生物难降解性和累积性[1],因此,美国环境总署(EPA)把具有致癌、致畸和致突变的16种PAHs确定为重点控制污染物。欧盟发布的2005/69/EC指令,对填充油和轮胎中8种PAHs的含量进行限制[2]。欧盟REACH法规附件X VII50也对消费品中的8种PAHs进行了限制[3]。德国认证技术文件ZEK 01.4-08[4]结合美国EPA及欧盟REACH法规管控的PAHs,将PAHs测试种类扩大到了18种。
丁基橡胶塞因其良好的气密性、低吸湿率、稳定性、耐老化性和低析出性而被广泛用于医疗器械生产和药品包装[5]。丁基橡胶塞在生产过程中需加入硫化剂、硫化促进剂、硫化活性剂、补强剂、填充剂、软化剂、防老剂、着色剂等多种加工助剂,以使橡胶基体获得良好的加工性能和使用性能[6,7],PAHs通常随填充油(软化剂)和炭黑(补强剂和着色剂)的使用而被引入其中[8]。丁基橡胶塞与药品长期接触,多环芳烃等有毒有害物质可能会迁移进入药品,这不仅会影响药品的质量,还有可能对患者的健康造成威胁。因此,需建立快速、准确、灵敏的方法测定药用丁基橡胶塞中多环芳烃的含量。
目前,针对橡胶类产品中PAHs的检测主要包括气相色谱-质谱法(GC-MS)、气相色谱-串联质谱法(GC-MS/MS)及高效液相色谱法(HPLC)[7,9 - 14]。但现有方法主要集中在15~17种PAHs的测定上,存在同分异构体不能有效分离的缺点。另外,由于橡胶类产品添加剂种类繁多、成分复杂,若样品浸提液不经净化,直接进样GC-MS分析会存在基质干扰,影响PAHs的定性及定量,并且容易造成进样口、色谱柱、检测器等污染,影响仪器的使用寿命[10]。本文采用超声浸提法对药用丁基橡胶塞中的PAHs进行极限浸提,通过固相萃取柱对样品浸提溶液进行净化,同时对浸提条件进行了优化。利用GC-MS法建立了药用丁基橡胶塞中18种PAHs的分析方法,并对不同生产厂家的样品中18种PAHs的含量进行了测定。该方法灵敏度高、准确性好,抗基质干扰能力强,适用于药用丁基橡胶塞中18种PAHs的检测。
1 实验部分
1.1 仪器与材料
7890A-5977B气相色谱-质谱仪(美国,安捷伦公司);Turbovap LV全自动氮吹浓缩仪(瑞典,Biotage公司)。PAH-MIP固相萃取柱(博纳艾杰尔,规格1 g/6mL);Strata©PAH固相萃取柱(飞诺美,规格750 mg/6mL);Oasis©HLB固相萃取柱(沃特世,规格5cc,200 mg);Oasis©HLB固相萃取柱(沃特世,规格6cc,200 mg)。
18种多环芳烃(表1)混合标准溶液(美国o2si公司,浓度:1 000 mg/L,溶剂为二氯甲烷),萘-D8标准溶液(美国o2si公司,浓度:2 019 mg/L,溶剂为二氯甲烷);蒽-D10标准品(德国Dr Ehrenstorfer GmbH公司,批号:G1001140,纯度:98.94%);苝-D12标准品(德国Dr Ehrenstorfer GmbH公司,纯度:99.42%)。分别取内标萘-D8、蒽-D10和苝-D12适量,用二氯甲烷配制成浓度约为2 μg/mL的混合内标溶液。标准曲线溶液:用二氯甲烷将18种PAHs标准品稀释成一定浓度的标准储备液,临用前精密吸取二氯甲烷1 mL,加入混合内标溶液20 μL、不同量的PAHs标准储备液,配制成浓度为1 ng/mL、2 ng/mL、4 ng/mL、5 ng/mL、6 ng/mL、8 ng/mL、10 ng/mL、20 ng/mL、50 ng/mL、100 ng/mL、150 ng/mL、200 ng/mL的标准曲线溶液(内标浓度约为40 ng/mL)。甲苯、乙酸乙酯、正己烷、二氯甲烷及丙酮均为色谱纯。
药用丁基橡胶塞样品均由企业提供。
1.2 样品的超声浸提
将橡胶塞样品剪成粒径为2 mm~3 mm的颗粒,称取1 g(精确至0.0001 g),置于20 mL顶空瓶中,加入5 mL正己烷-丙酮(体积比1∶1),再精确加入混合内标溶液20 μL,混匀,密闭。在60 ℃水浴下超声1 h。待顶空瓶冷却至室温后,将浸提液转移至玻璃试管中。再向顶空瓶中加入5 mL正己烷-丙酮混合溶液,重复上述操作2次,将3次的浸提液合并至玻璃试管中,待净化。
1.3 萃取溶液净化
将浸提液于35 ℃水浴中氮吹至近干,用2 mL正己烷复溶,过固相萃取柱(预先依次用20 mL二氯甲烷和10 mL正己烷活化),再用2 mL正己烷冲洗玻璃试管,重复操作两次,将冲洗液过柱,弃去上述滤液。用10 mL二氯甲烷淋洗,收集淋洗液。将淋洗液在35 ℃水浴中氮吹浓缩至1 mL,涡旋振荡混匀,如有必要过0.22μm有机滤膜,待测。
1.4 气相色谱-质谱条件
气相色谱条件:DB-EUPAH色谱柱(60 m×0.25 mm×0.25 μm);进样口温度:300 ℃;进样量:1 μL;不分流;流速:1.4 mL/min;柱温:60 ℃保持2 min,以25 ℃/min升温至200 ℃,再以20 ℃/min升温至320 ℃并保持24 min;传输线温度:300 ℃。
质谱条件:离子源类型:电子轰击离子源(EI)源;离子源温度:230 ℃;MS四极杆温度:150 ℃;溶剂延迟:7.5 min;扫描方式:选择离子模式(SIM)。
2 结果与讨论
2.1 色谱柱的选择
根据相关文献报道[11 - 13,15],在采用GC-MS或GC-MS/MS法测定PAHs含量时,通常选用DB-EUPAH和HP-5MS这两种色谱柱。考察了HP-5MS(60 m×0.25 mm×0.25 μm)和DB-EUPAH(60 m×0.250 mm×0.25 μm)两种色谱柱。结果发现,在使用HP-5MS色谱柱时,同分异构体苯并(b)荧蒽、苯并(k)荧蒽和苯并(j)荧蒽无法完全分离,使用DB-EUPAH色谱柱时,在优化后的色谱条件下,18种PAHs可以完全分离,色谱见图1。萘-D8与萘、菲和蒽-D10在色谱图中存在的峰重叠现象,可通过它们的定性、定量离子的不同来区分目标物与内标。
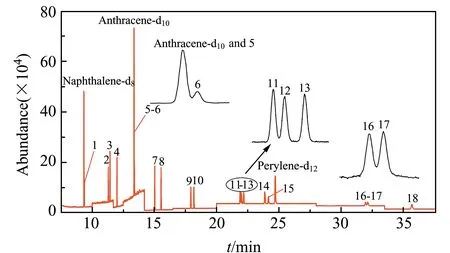
图1 PAHs和内标的选择离子色谱图Fig.1 Selected ion chromatograms of PAHs and internal standard compounds
2.2 样品前处理条件的优化
2.2.1 固相萃取柱的选择根据文献报道[1,9,10],PAHs大多采用硅胶固相萃取柱进行净化,但董彩玉等[9]报道过柱液重现性较差。因此,我们实验了市售固相萃取柱的净化效果。发现其中两家的固相萃取柱虽然对样品浸提液有净化作用,但对分子量较小的PAHs的回收率低。而博纳艾杰尔的固相萃取柱不仅能对样品浸提溶液起到净化作用,降低背景干扰,还可确保目标物在净化过程中不损失。将样品浸提液、正己烷上样滤液和二氯甲烷淋洗液分别用二氯甲烷定容至10 mL,然后分别用全扫描模式(SCAN,扫描范围m/z45~450)和SIM模式进行分析。结果表明,样品浸提溶液在用上述固相萃取柱净化过程中,其中的杂质在用正己烷上样时会被保留在萃取柱中,但经固相萃取柱净化后,基质干扰极大降低,不仅可避免假阳性结果的出现,还有助于样品中目标物的准确定量。
2.2.2 浸提溶剂的选择参照相关文献报道[7,9,10],选用乙酸乙酯、正己烷、甲苯、正己烷-二氯甲烷(1∶1)、正己烷-乙酸乙酯(1∶1)、正己烷-丙酮(1∶1)6种常见的PAHs浸提溶剂,通过对1个阳性样品进行浸提,考察不同浸提溶剂的浸提能力。结果表明,样品中含有苊、芴、菲、荧蒽以及芘,乙酸乙酯浸提的峰响应值最大,记为100%,其次为正己烷-丙酮(1∶1),其所得样品溶液中5种目标物的峰响应相对值均在95%以上,与乙酸乙酯几乎相同,其余4种溶剂对目标物的浸提效果明显比前述两种溶剂差,目标物的峰响应相对值大部分在80%以下。考虑到正己烷-丙酮(1∶1)氮吹至近干耗时短、所需水浴温度低,有利于提高样品前处理效率,因此选择正己烷-丙酮(1∶1)为浸提溶剂。
2.2.3 浸提比例的优化采用正己烷-丙酮(1∶1)为浸提溶剂,按不同浸提比例,对1个阳性样品进行浸提。具体操作如下:称取样品1 g(精确至0.0001 g),分别加入5 mL、10 mL、15 mL和20 mL正己烷-丙酮(1∶1),密封,于60 ℃水浴中超声1 h,得浸提液,按仪器条件进样分析,记录峰面积。结果表明,样品中含有苊、芴、菲、荧蒽以及芘,随着浸提比例的增大,其响应值有所增加,但增幅仅在10%左右,当浸提溶剂体积为20 mL时,浸提出的多环芳烃的响应值反而下降。综合考虑,最终确定浸提比例为5 mL/g。
2.2.4 极限浸提考察称取样品1 g(精确至0.0001 g),加入5 mL正己烷-丙酮(1∶1),密封,于60 ℃水浴中超声1 h,制得样品浸提液。然后,再向上述样品中加入5 mL正己烷-丙酮(1∶1),重复上述操作直至达到极限浸提。结果表明,第二次浸提时各检出的PAHs的峰面积约为首次浸提检出量的30%,第三次浸提时各检出的PAHs的峰面积约为首次浸提检出量的10%。因此,选择样品超声浸提3次,合并浸提液作为样品浸提液。
2.3 方法学考察
2.3.1 检出限和定量限取混合标准溶液进样分析,按信噪比(S/N)为3时的浓度为检出限,S/N为10且具有良好精密度和准确度的浓度为定量限。本方法18种PAHs的检出限和定量限见表1。结果显示,萘检出限为4.0 ng/mL,定量限为10.0 ng/mL;二苯并(a,h)蒽检出限和定量限均为10.0 ng/mL;其余PAHs的检出限为0.2~1.0 ng/mL,定量限为1.0~2.0 ng/mL,表明方法检出限良好,灵敏度高。
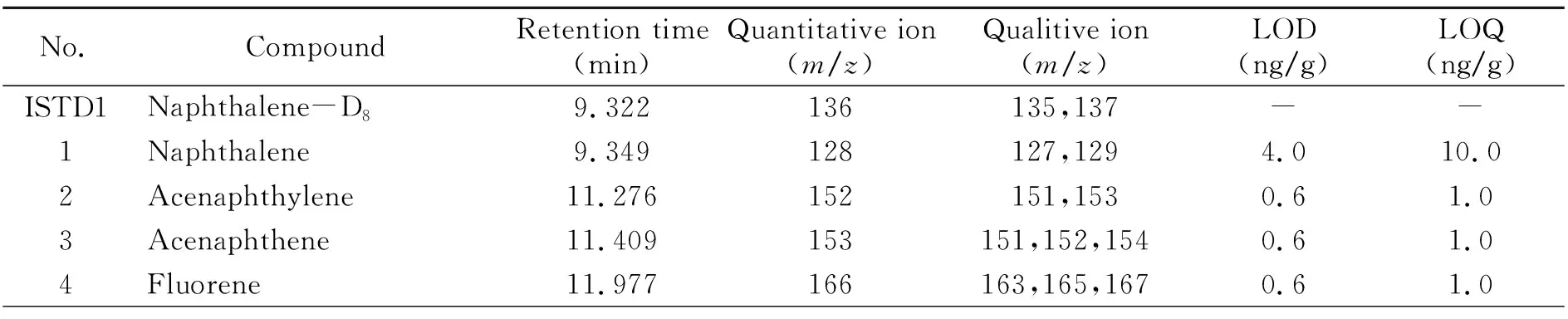
表1 18种PAHs和内标的保留时间、定量离子、定性离子、检出限(LOD)和定量限(LOQ)Table 1 Retention times,quantitative ions,qualitive ions,LODs and LOQs of 18 PAHs and internal standards(ISTDs)
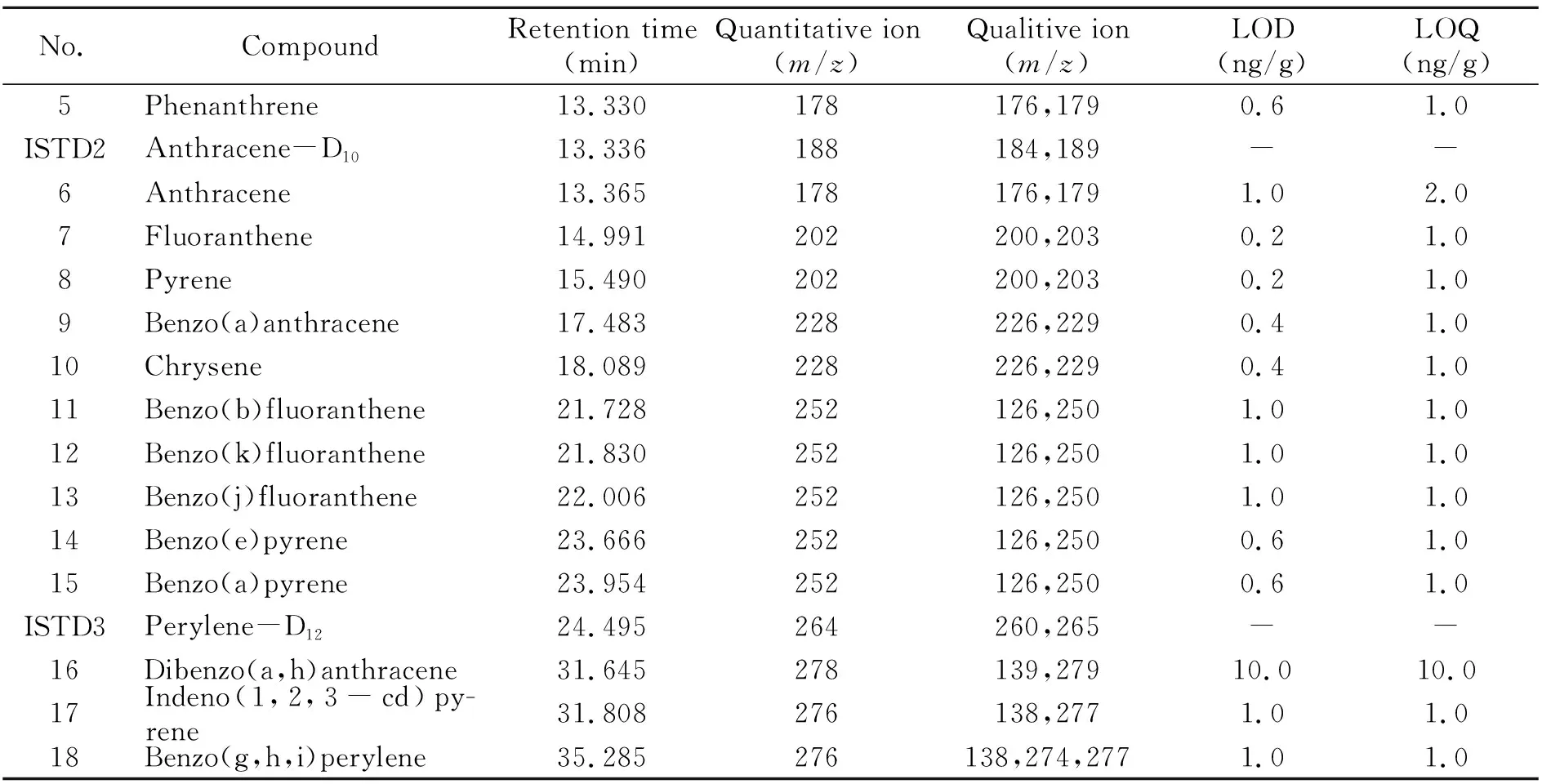
(续表1)
2.3.2 线性和范围采用内标法绘制标准曲线,见表2。结果显示,在1~10 ng/mL和10~200 ng/mL范围内,所有目标物均呈现良好的线性关系,线性相关系数r均大于0.9992。
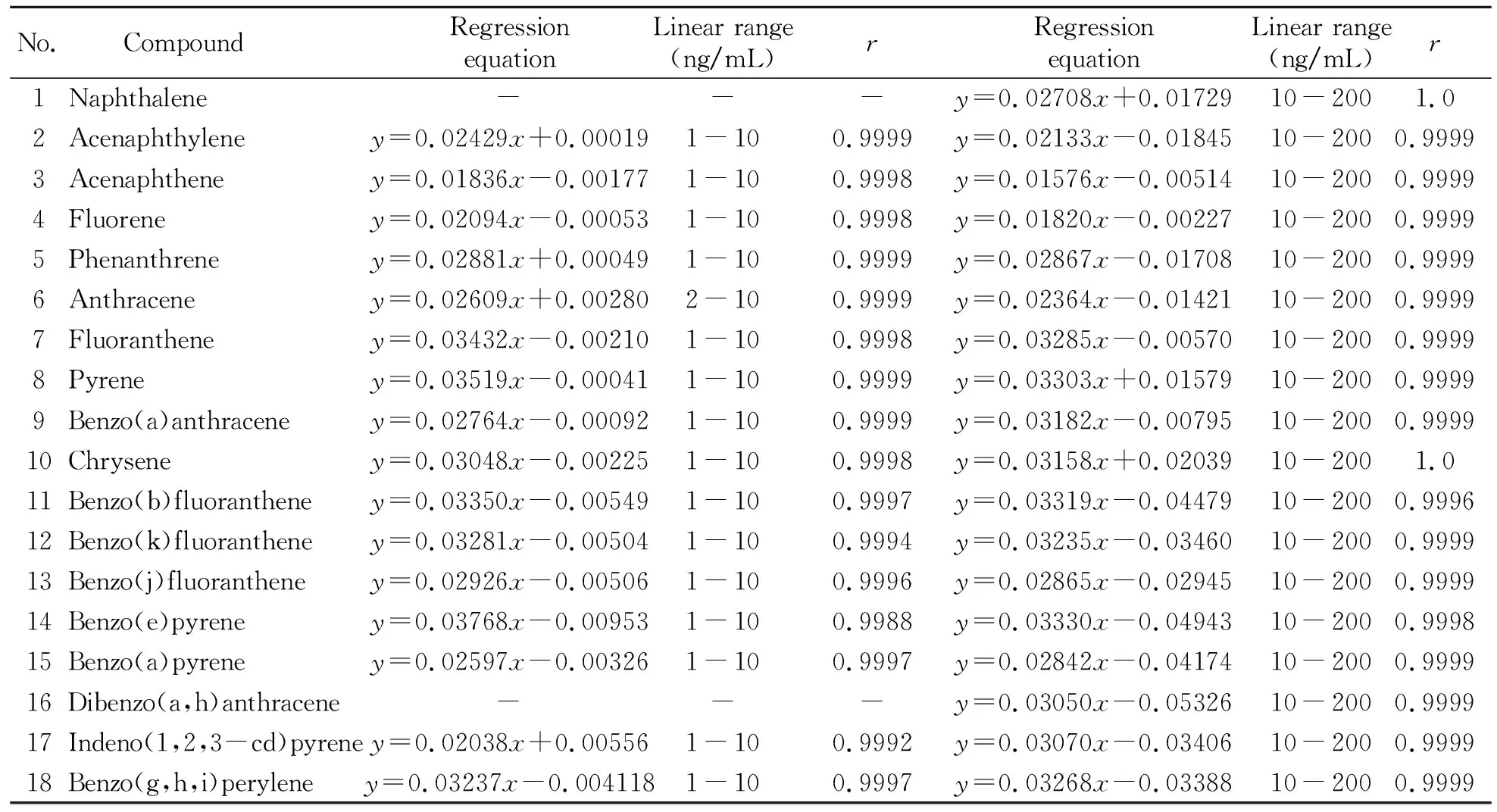
表2 PAHs的线性范围、线性方程和相关系数Table 2 Regression equations,linear ranges and correlation coefficients(r) of PAHs
2.3.3 精密度和回收率取浓度为1 ng/mL、10 ng/mL和100 ng/mL的混合标准溶液分别连续进样6次,计算相对标准偏差(RSD)。在上述3个浓度水平进行样品加标回收试验中,每个浓度水平做3个平行样,结果见表3。18种PAHs的加标回收率为76.63%~113.93%,RSD值为0.11%~6.86%。上述结果表明方法精密度和准确度良好,能够满足分析要求。
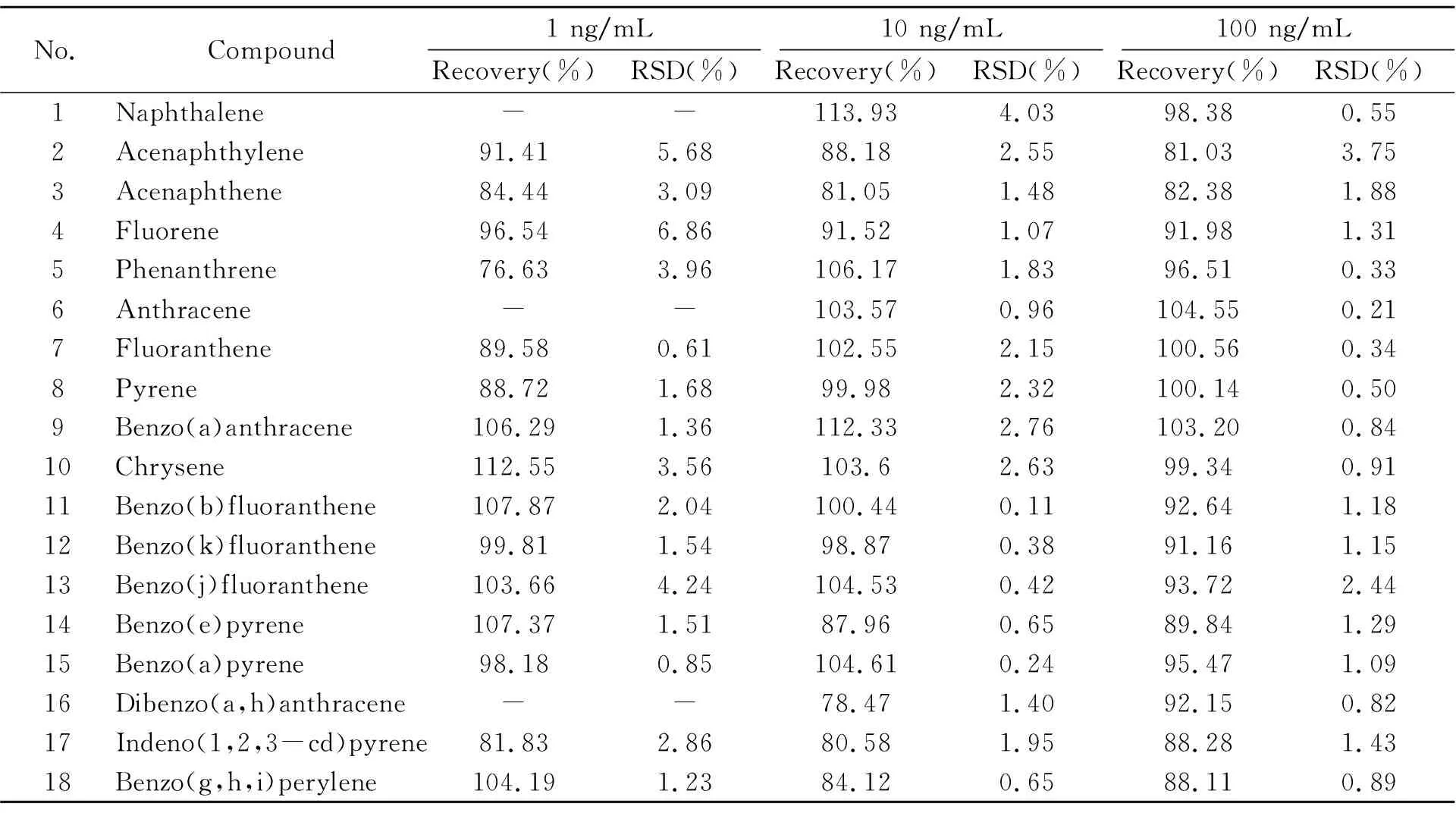
表3 PAHs的精密度和加标回收率(%)Table 3 Precisions and recoveries of PAHs(%)
2.4 药用丁基胶塞样品的检测

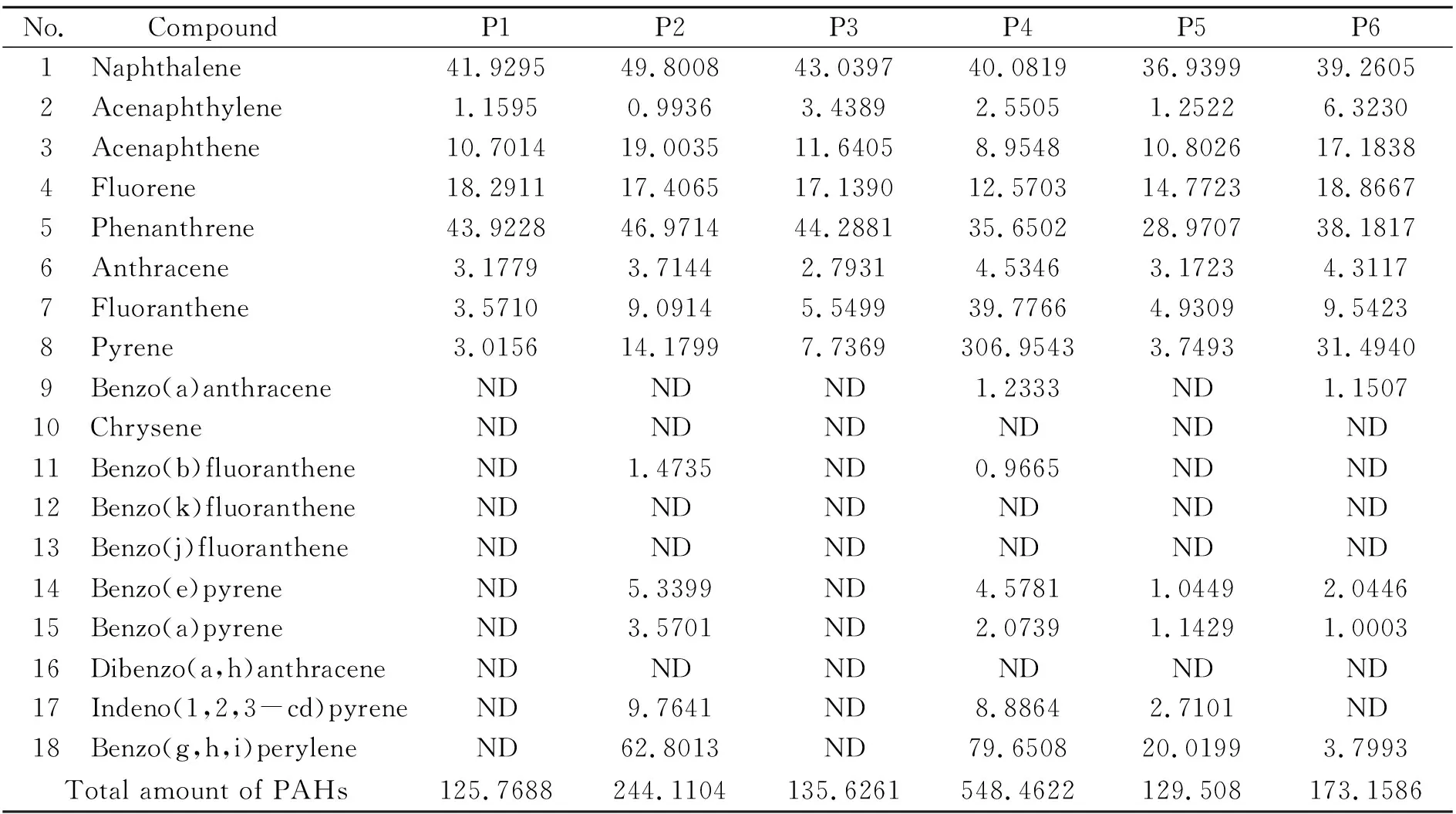
表4 不同厂家生产的药用丁基胶塞中18种PAHs 的含量(ng/g)Table 4 Contents of PAHs in pharmaceutical butyl rubber closures acquired from different producers (ng/g)
3 结论
本文建立了GC-MS法测定药用丁基橡胶塞中18种PAHs含量。该方法灵敏度高、准确度好,能够有效降低胶塞样品中的基质干扰,避免假阳性结果的出现,适用于药用丁基胶塞中18种PAHs含量的测定,为此类产品的安全评价和质量控制提供了方法参考。
