衍生化-分散液液微萃取-气相色谱/质谱法同时测定纺织固体废物中含氯苯酚和邻苯基苯酚
2021-11-15铁建成龙志新贾丽霞孙慧芹藏蒙蒙
铁建成, 龙志新, 刘 俊, 贾丽霞,朱 鹏, 孙慧芹, 藏蒙蒙
(1.新疆大学纺织与服装学院,新疆乌鲁木齐 830046;2.成都海关技术中心,四川成都 610041;3.乌鲁木齐海关,新疆乌鲁木齐 830063)
纺织固体废物来源广泛、物质复杂且毒性物质潜在风险严重,给我国生态环境安全带来了严重威胁。一氯苯酚(CP)、二氯苯酚(DCP)、三氯苯酚(TrCP)、四氯苯酚(TeCP))和邻苯基苯酚(OPP)常被用于纺织品的杀虫剂、防霉防蛀剂及消毒剂[1 - 3],是常见的几种有害物质。他们结构稳定,溶解度大,降解性能极差,土壤、水体、固体废物等对其吸附和固定作用很弱,在环境和生物体内易产生富集作用[4]。因其对人体具有致突变性、致癌性和致畸性,因此被美国环境保护署和其他国家列为优先污染物行列[5 - 9]。中国的生态纺织标准和国际生态纺织协会的Oeko -tex标准100,均明确规定纺织品中TeCP的残留量不得超过0.5 mg/kg,婴幼儿用品不得超过0.05 mg/kg[10]。目前,对含氯苯酚的测定已有标准方法[11 - 14],但这些方法均存在有机溶剂消耗量大、萃取时间长、灵敏度低等问题,且检测物质过于单一,无法同时满足对痕量CP、DCP、TrCP、TeCP和OPP的同时测定。
分散液液微萃取(DLLME)具有操作简单、富集倍数高、有机溶剂用量少、成本低廉等优点,因此得到了广泛应用[15 - 20]。目前,关于纺织固体废物中CP、DCP、TrCP、TeCP和OPP的同时测定还未见发布相关标准方法。本文采用衍生化-分散液液微萃取前处理技术,结合气相色谱/质谱(GC/MS)检测方法,实现了同时对纺织固体废物中18种含氯苯酚和邻苯基苯酚的操作简便、有机溶剂用量少、成本低廉、灵敏度较高和准确性较好的测定方法,为相关检测部门对大批量样品的检测需求提供一定的技术支持。
1 实验部分
1.1 主要仪器与试剂
Agilent 7890A/5975C气相色谱/质谱联用仪(美国,Agilent公司);KQ-300DE型数控超声波清洗器(昆山市超声仪器有限公司);N-EVAP 112水浴氮吹仪(美国,Organomation公司);电子天平(瑞士,梅特勒-托利多);SmarVapor RE 501旋转蒸发仪(德国,De Chem-Tech公司);Allegra X-22R高速离心机(德国,Beckman公司);EYELA MMV-1000W振荡器(日本,东京理化公司);MS 3型涡旋混匀器(德国,IKA公司)。
标准品:2-氯苯酚(2-CP)、3-氯苯酚(3-CP)、4-氯苯酚(4-CP)、2,3-二氯苯酚(2,3-DCP)、2,4-二氯苯酚(2,4-DCP)、2,5-二氯苯酚(2,5-DCP)、2,6-二氯苯酚(2,6-DCP)、3,4-二氯苯酚(3,4-DCP)、3,5-二氯苯酚(3,5-DCP)、2,3,4-三氯苯酚(2,3,4-TrCP)、2,3,5-三氯苯酚(2,3,5-TrCP)、2,3,6-三氯苯酚(2,3,6-TrCP)、2,4,5-三氯苯酚(2,4,5-TrCP)、2,4,6-三氯苯酚(2,4,6-TrCP)、3,4,5-三氯苯酚(3,4,5-TrCP)、2,3,4,5-四氯苯酚(2,3,4,5-TeCP)、2,3,4,6-四氯苯酚(2,3,4,6-TeCP)、2,3,5,6-四氯苯酚(2,3,5,6-TeCP)、邻苯基苯酚(OPP),购自德国Dr.Ehrenstorfer GmbH公司,纯度均大于98.0%。甲醇、丙酮、异丙醇、氯苯、三氯甲烷、乙腈、四氯化碳均为色谱纯(德国Merck公司);二氯甲烷(色谱纯,Fisher公司);乙酸酐、K2CO3、NaOH、NH3·H2O、无水乙醇、无水Na2SO4(450 ℃烘6 h)均为分析纯(天津致远化学试剂有限公司)。实验用水由ELIX5+60L纯水机(美国密理特公司)制备。
样品:市场委托和进口报检的62批次纺织固废物,包含废棉(粗疏和精梳落棉、絮胎、棉短绒)、废棉纱线、废布样(废棉布料、废牛仔布料、废涤棉混纺布料)。
1.2 样品前处理
1.2.1 提取取代表性纺织固体废物样品,裁剪成3 mm×3 mm,准确称取样品1 g(精确至0.01 g),加入15 mL 0.15 mol/L K2CO3溶液,于磁力搅拌器上搅拌5 min,使样品与溶液充分混合,室温条件下超声萃取15 min,过滤。残留物按以上步骤重复萃取一次,合并两次提取液,定容至30 mL。
1.2.2 衍生与DLLME萃取准确移取6 mL样品提取液于10 mL尖底离心管中,加入0.12 mL乙酸酐,拧紧瓶盖,涡旋混匀,于60 ℃条件下衍生35 min后,冷却至室温,将0.7 mL体积比为2∶5的四氯化碳(萃取剂)和丙酮(分散剂)的混合溶剂迅速注入到尖底离心管中,涡旋混匀,于常温40 kHz条件下超声萃5 min,在8 000 r/min下离心3 min,抽取下层有机相进行GC/MS分析。
1.3 GC/MS条件
采用HP-5MS毛细管色谱柱(30 m×0.25 mm,0.25 μm),进样口温度280 ℃,载气:高纯氦气(99.999%),流速1.0 mL/min;程序升温:50 ℃保持2 min,以10 ℃/min升至230 ℃,不分流进样,进样量1.0 μL。传输线温度:280 ℃,离子源温度:230 ℃,电离方式:电子轰击离子源(EI),电离能量:70 eV,选择离子监测(SIM)模式检测,质量扫描范围:45~500 amu,电子倍增器电压:1.4 kV,溶剂延迟:8 min。全部程序总时长为20 min。
1.4 定性依据及定量方法
根据色谱峰的保留时间、特征离子、定量及定性离子的相对丰度比进行定性。对样品进行测定时,如果试样中的质量色谱保留时间与标准工作溶液中的色谱峰保留时间一致(变化范围在±0.5%之间),在扣除背景后的质谱图中所选特征离子均出现,且相对丰度与浓度相当的标准工作溶液的相对丰度相差不大于15%,则可以判断样品中含有该目标物质。采用外标法进行定量。18种含氯苯酚和邻苯基苯酚的保留时间、定量离子、定性离子及相对丰度比见表1。

表1 18种含氯苯酚和邻苯基苯酚的保留时间、特征离子及相对丰度比Table 1 Retention times,characteristic ions and relative abundance ratios of 18 chlorophenols and o -phenylphenols
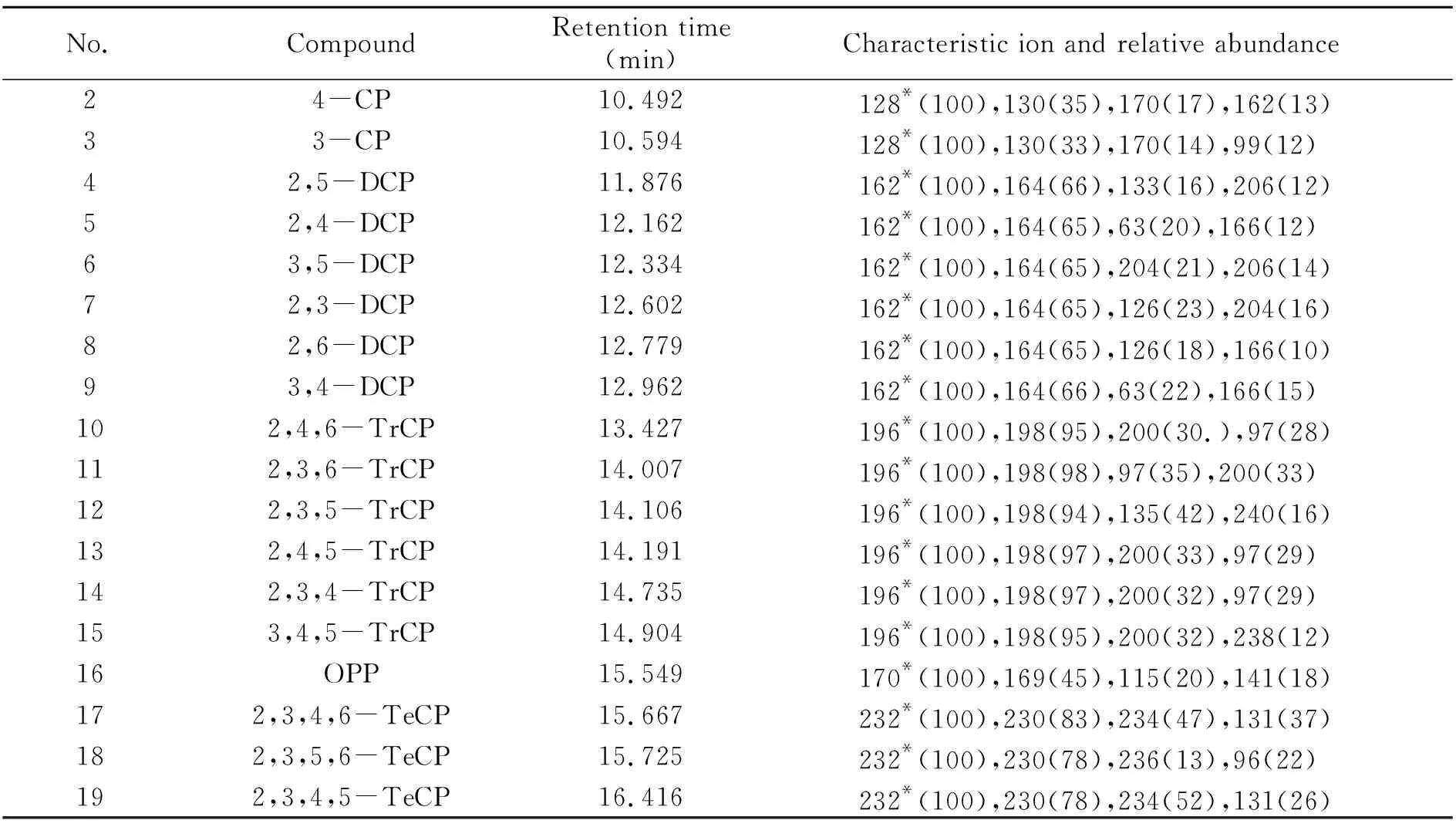
(续表1)
1.5 工作曲线的绘制
将18种含氯苯酚和邻苯基苯酚标准品分别用甲醇配制成1 000 mg/L的标准储备溶液,置于棕色储存瓶中,于-20 ℃条件下避光储存。根据需要用丙酮配制成10 mg/L的混合标准工作溶液,置于棕色储存瓶中,于-20 ℃条件下避光储存。取7份0.15 mol/L K2CO3溶液进行加标,加标后18种含氯苯酚和邻苯基苯酚的浓度分别为0.002、0.005、0.01、0.02、0.04、0.08、0.16 mg/L,各取6 mL按照“1.2.2”进行衍生化处理后进行测定,绘制标准曲线。
2 结果与讨论
2.1 18种含氯苯酚和邻苯基苯酚乙酰化后DLLME色谱图
根据“1.2”前处理步骤和“1.3”仪器条件进行实验,所得标准色谱图如图1所示。18种含氯苯酚和邻苯基苯酚萃取效率良好,均可以实现较好的分离,符合分析检测的要求。
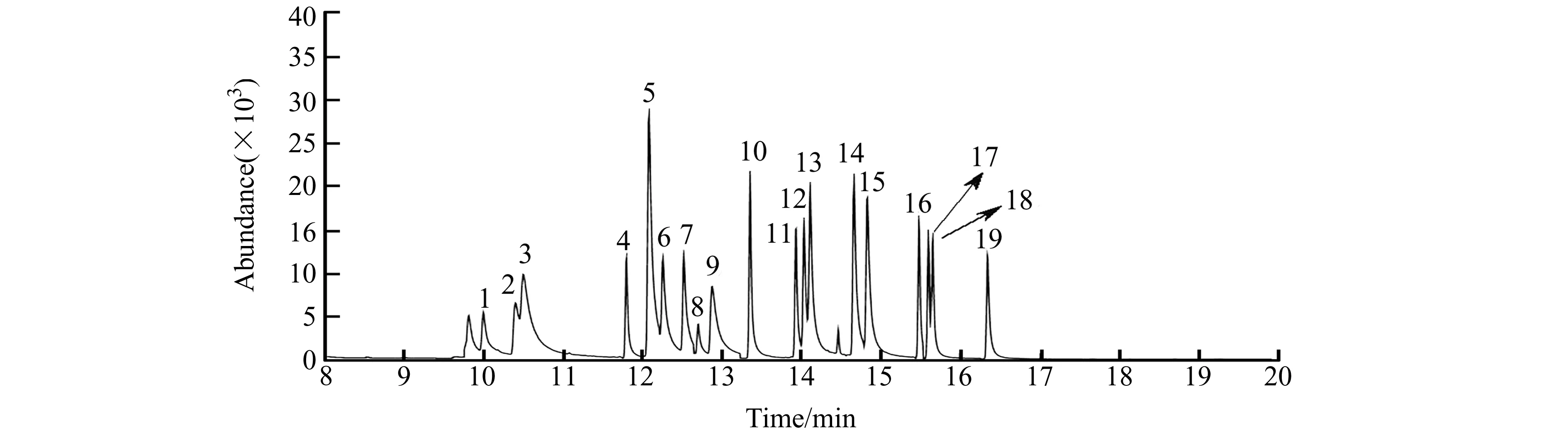
图1 含氯苯酚和邻苯基苯酚混合标准溶液乙酰化后DLLME色谱图Fig.1 Chromatogram of DLLME after acetylation of mixed standard solutions for chlorophenols and o -phenylphenols1 - 19 refer to the 19 compounds in Table 1.
2.2 萃取体系的选择和优化
2.2.1 萃取基体的选择通常含氯苯酚类化合物呈弱酸性,因此以弱碱性溶剂提取可以使其以离子状态存在于溶液中,增加溶解度,从而提高萃取效率。实验分别考察了不同浓度的NaOH溶液、NH3·H2O和K2CO3溶液作为萃取溶剂时对目标物的提取效率。取具有代表性的2,5-DCP、2,3,4-TrCP、2,3,4,5-TeCP、OPP作为目标化合物,添加到阴性样品中,按照“1.2”前处理方法和“1.3”仪器条件进行测试。结果表明,碱性溶液在0.01~0.15 mol/L范围内,提取效率随浓度的增加而增加,当浓度为0.15 mol/L时提取效率达到最大,再增加浓度,提取效率反而出现下降的趋势(图2),原因可能是强碱性环境对化合物以及仪器响应都产生了一定的影响。当碱性溶液浓度都达到0.15 mol/L时,K2CO3溶液的提取效率明显优于NaOH溶液和NH3·H2O。因此,本文选择浓度为0.15 mol/L K2CO3溶液作为18种含氯苯酚和邻苯基苯酚萃取溶剂。

图2 不同浓度K2CO3溶液(a)、NaOH溶液(b)和弱碱性NH3·H2O(c)作为萃取溶剂对2,5-DCP、2,3,4-TrCP、2,3,4,5-TeCP和OPP提取效率的影响Fig.2 The effect of different concentrations of K2CO3 solution(a),NaOH solution(b) and weak alkaline NH3·H2O(c) as extraction solvents on the extraction efficiency of 2,5-DCP,2,3,4-TrCP,2,3,4,5-TeCP and OPP
2.2.2 衍生试剂用量、温度与时间的优化在0.15 mol/L的K2CO3溶液中加入0.8 mg/L标准工作溶液,以四氯化碳作为萃取剂,丙酮作为分散剂,考察了衍生试剂乙酸酐由0.01 mL增加到0.3 mL对实验的影响。结果显示,当乙酸酐用量在0.08~0.12 mL之间时,峰面积有明显的增加,当体积超过0.15 mL时,峰面积出现一定程度的下降,可能的原因是过量的乙酸酐影响了溶液体系的pH,同时乙酸酐过量会对色谱柱产生一定的影响。实验最终选择衍生试剂乙酸酐的用量为0.12 mL。
含氯苯酚属于易挥发物质,反应温度会对衍生化过程产生一定的影响,因此在保证反应温度使含氯苯酚类物质有较大的溶解度的同时降低其挥发性。本实验在35~80 ℃内确定的较为理想的衍生温度为60 ℃。衍生时间应保证体系内的酚类物质与乙酸酐充分反应,当反应时间达到一定条件后,继续延长反应时间,大部分目标物质的峰面积无明显变化,个别峰面积有所下降,可能的原因是由于酯的水解所导致。因此本实验在20~60 min内确定较为理想的衍生时间为35 min。
2.2.3 萃取剂与分散剂的选择以丙酮作为分散剂,分别考察了氯苯、三氯乙烯、四氯乙烯、四氯化碳作为萃取剂时对各目标组分的萃取效率。结果如图3所示,当以四氯化碳作为萃取剂时,各个目标物质的萃取效率相对较好,富集倍数较为理想,且四氯化碳对目标组分无干扰,故选用四氯化碳作为本实验的萃取溶剂。
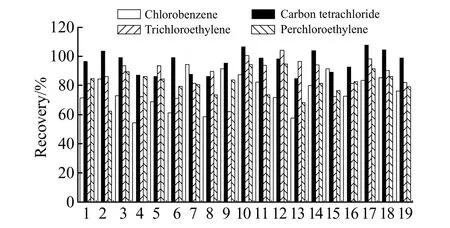
图3 氯苯、二氯甲烷、三氯甲烷、四氯化碳作为萃取剂时各目标组分的萃取效率Fig.3 Extraction efficiency of each target component when chlorobenzene,dichloromethane,chloroform,and carbon tetrachloride are used as extractants1 - 19 refer to the 19 compounds in Table 1.
以四氯化碳作为萃取剂,分别考察了丙酮、异丙醇、乙腈、甲醇作为分散剂时对目标组分提取效率的影响。实验结果显示,当以丙酮作为分散剂时,各个目标物质的富集倍数较好,各个组分的萃取效率均高于其他3种分散剂,且相对标准偏差(RSD)相对较低,故选择丙酮作为分散剂。
2.2.4 萃取剂与分散溶剂用量的优化以0.4 mL丙酮作为分散剂,分别考察了0.1、0.2、0.3、0.4 mL的四氯化碳对萃取效率的影响。结果表明,随着四氯化碳体积的增加萃取效率呈现出明显下降的趋势,考虑到萃取剂体积过小时不利于取液进样,因此本实验选择萃取剂的理想用量为0.2 mL。以0.2 mL四氯化碳作为萃取剂,分别考察了四氯化碳和丙酮不同体积比时对目标化合物提取效率的影响。结果如图4所示。当四氯化碳和丙酮体积比为2∶5时,除3,5-DCP、2,3,6-TrCP 和3,4,5-TrCP外其余目标化合物的萃取率均高于其他体积比。综合考虑,最终选择萃取剂和分散剂的体积比为2∶5,用量为0.7 mL。
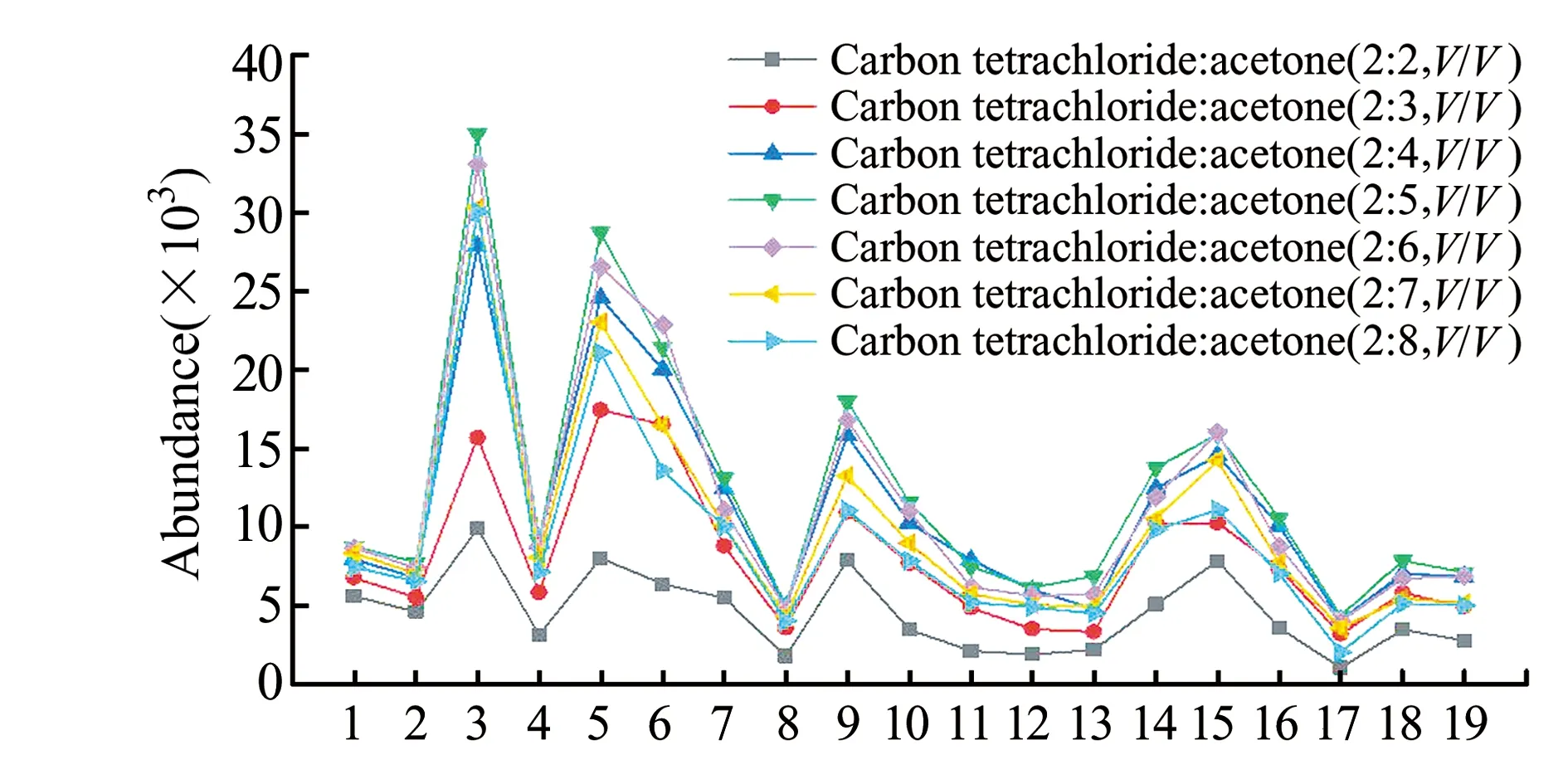
图4 不同体积比的四氯化碳和丙酮对萃取效率的影响Fig.4 The effect of different volume ratios of carbon tetrachloride and acetone on extraction efficiency1 - 19 refer to the 19 compounds in Table 1.
2.3 标准曲线、线性范围和检出限
配制7个不同浓度(0.002、0.005、0.01、0.02、0.04、0.08、0.16 mg/L)的混合标准工作溶液,以混合标准工作溶液的质量浓度(x)为横坐标,定量离子色谱峰面积(y)为纵坐标,绘制标准工作曲线,得到线性方程和相关系数,结果见表2。各目标物质在0.002~0.16 mg/L范围内呈现出良好的线性关系,相关系数为0.9991~1.0000。根据HJ 168-2020《环境监测分析方法标准制修订技术导则》关于检出限的计算方法,对浓度值为估计方法检出限值2~5的样品进行7次平行测定,计算各个目标物质的检出限,并进行验证。最终确定18种含氯苯酚和邻苯基苯酚的检出限(LOD)为0.07~0.76 μg/kg,以4倍检出限来限定其定量限(LOQ)为0.28~3.04 μg/kg。

表2 18种含氯苯酚和邻苯基苯酚的线性方程、线性范围、线性相关系数(r)、检出限(LOD)和定量限(LOQ)Table 2 Linear equation,linear range,linear correlation coefficient(r),limit of detection(LOD) and limit of quantification(LOQ) of 18 chlorophenols and o -phenylphenols
2.4 加标回收率和精密度
取代表性纺织固体废物样品,分别添加3个不同浓度(0.005、0.04、0.1 mg/L)的加标水平,按照“1.2”前处理方法对样品进行处理后,按“1.3”仪器条件上机测试,每一个加标水平做6次平行,结果见表3。18种含氯苯酚和邻苯基苯酚加标回收率为84.2%~105.0%,相对标准偏差(RSD)为0.6%~6.4%。

表3 不同纺织固体废物样品中酚类化合物的检测结果Table 3 Test results of phenolic compounds in different textile solid waste samples
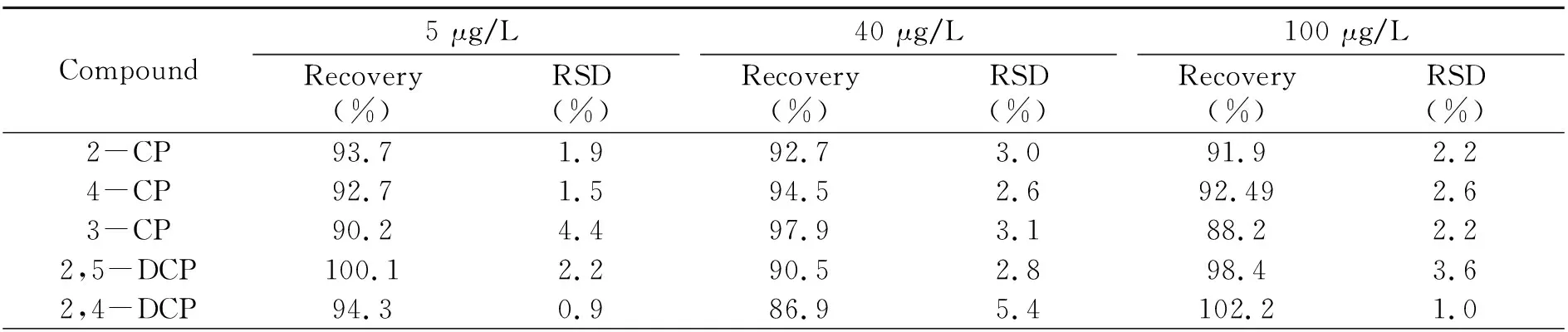
表3 18种含氯苯酚和邻苯基苯酚加标回收率和相对标准偏差(RSD)Table 3 Spiked recovery and relative standard deviation(RSD) of 18 kinds of chlorinated phenols and o -phenylphenols

(续表3)
2.5 与标准方法的比较
从前处理方法、萃取溶剂和用量、检出限和回收率等方面,分别对国际标准委员会制定的标准(ISO 17070:2006)[11],我国国家标准(GB/T 24166-2009)[12]、(GB/T 18414.2-2006)[13]、(GB/T20386-2006)[14],与本文所建立的DLLME-GC/MS方法进行了比较。本方法实现了基质更为复杂的纺织固体废物中18种含氯苯酚和邻苯基苯酚的测定,方法具有简单、有机溶剂用量少、成本低廉、灵敏度较高、准确性较好等优点。
2.6 实际样品分析
采用本文建立的方法对市场委托和进口报检的62批次纺织固废物进行18种含氯苯酚和邻苯基苯酚的测定,结果见表5。
3 结论
本文建立了纺织固体废物中18种含氯苯酚和邻苯基苯酚的同时测定方法,对提取液中目标物质乙酰化后,采用液液微萃取对衍生产物进行提取,气相色谱-质谱进行检测,讨论并优化了实验最佳的条件。结果证明,含氯苯酚类物质通过衍生化以后,峰形得到了显著的改善,方法的检出限降低,使定性和定量分析更加精确化。衍生化技术和分散液液微萃取(DLLME)技术相结合,用于测定纺织固体废物中18种含氯苯酚和邻苯基苯酚是一种具有操作简单、有机溶剂用量少、成本低廉、灵敏度较高、准确性较好测定方法。
