基于SLAF-BSA技术的蝴蝶兰花底色关联SNP分子标记开发与验证
2021-11-09肖文芳陈和明吕复兵
肖文芳 李 佐 陈和明 吕复兵
(广东省农业科学院 环境园艺研究所/广东省园林花卉种质创新综合利用重点实验室,广州 510640)
蝴蝶兰属(Phalaenopsis)为兰科商品化程度最高的属,具极高的观赏价值和商业价值,是全球最重要的观赏花卉之一。花色是观赏花卉最重要的性状评价指标之一,直接影响其观赏价值和商业价值[1]。有别于绝大多数观赏花卉的单色花和少数复色花,蝴蝶兰的花瓣和萼片具丰富的颜色分布类型,除匀色花外,绝大多数种质资源的花瓣和萼片为双色,可分为底色和斑色,一般底色为整朵花的主色,斑色的颜色分布类型则包括阴影、镶边、条纹、线纹、网纹、斑点、斑块、阴影和镶边、线纹和斑点、镶边和线纹、镶边与线纹和斑点[2]。植物花色属于复杂的数量性状,受多基因控制,遗传基础复杂[3]。蝴蝶兰原生种众多,商品品种经过一百多年的杂交选育,血统复杂,与原生种参考基因组的比对效率极低,且花色调控机制极为精细,通过转录组和简化基因组测序挖掘性状相关基因和关联分子标记仍然是目前研究商品品种花色遗传机制的重要手段[1,4],但尚未开发出花色相关分子标记。
特异位点扩增片段测序(Specific-locus amplified fragment sequencing,SLAF)是一种根据物种个性化设计方案的简化基因组测序技术,能够获取全基因组范围内的多态性标签并快速准确筛选性状关联单核苷酸多态性(Single-nucleotide polymorphism,SNP)。群分法(Bulked segregant analysis,BSA)与SLAF的结合,能够进一步加快对复杂性状定位的速度,目前已在水稻黄叶转绿基因定位[5]、玉米果皮纤维素含量相关基因定位[6]、大豆酸性磷酸酶活性候选基因挖掘及功能标记开发[7]、大豆半矮秆基因定位[8]、红麻第一花节位置相关SNP筛选[9]、辣椒果实花青素积累相关基因定位[10]和紫薇矮化性状相关SNP标记筛选[11]等研究得到成功运用。SLAF-BSA技术目前在兰科植物上还没有应用,但Lu等[12]单独利用SLAF技术成功构建了一张包含了8 573个SLAF标签的石斛兰高密度遗传图谱,并筛选出5个与石斛多糖含量相关的数量性状座位(Quantitative trait locus,QTL);Wang等[13]利用SLAF技术对文心兰长期继代培养中的体细胞无性系变异进行鉴定和筛选,证明SLAF技术在兰科植物上适用。
蝴蝶兰花底色直接决定整株花的主色调,且在传统杂交育种过程中发现底色与斑色是互不干扰的2个独立遗传性状。细化花色这一复杂性状,针对性地筛选与底色相关的分子标记,去除斑色的干扰,能够更精准获取与单个性状相关联的分子标记,为辅助育种提供更精确有效的筛选依据。本研究基于SLAF-BSA技术,对2个亲本和杂交F1代构建的2个极端性状混池(黄色底色混池和白色底色混池)DNA文库进行简化基因组测序,与花底色性状进行关联分析筛选相关标记位点,并在F1代群体及其他种质资源中进行验证,为蝴蝶兰分子标记辅助育种及花色改良基因工程提供理论支持。
1 材料与方法
1.1 试验材料
以‘黄金豹’蝴蝶兰(Phal.Frigdaas Oxford)为母本,‘白天使’蝴蝶兰(Phal.Join Angel)为父本构建杂交群体,共获得505个杂交后代,黄色底色个体与白色底色个体的比例为273∶232。后代可根据底色和斑色分为11个组(图1),性状分离具体信息详见李佐等的研究[14]。从Group 1、3、5、7和9这5个黄色底色群体中随机选取33个单株加上Group 11中的2个单株组成黄色底色混池(ab),从Group 2、4、6、8和10这5个白色底色群体中随机选取35个单株组成白色底色混池(aa),其中Group 11中的2个单株对应斑纹最少的Group 4。所有实验材料均栽培保存于广东省农业科学院环境园艺研究所白云基地兰花资源圃中,于2018年采用统一孔径打孔器分别等量采集样品倒数第二片叶顶端中部的叶片组织,4 ℃保存备用。

图1 杂交亲本及F1代个体代表株系的花部Fig.1 Flower characters of 11 Phalaenopsis F1 offspring groups and the parents
1.2 试验方法
1.2.1主要试剂及仪器
主要试剂:SNaPshot相关 PRISM®SNaPshotTM Multiplex Kit试剂盒购自Applied Biosystems公司;TaqHotStart DNA Polymerase购自Kapabiosystems公司;SAP酶购自Fermentas公司;ExoI酶、RsaI酶、HaeⅢ酶和CIP酶购自New England Biolabs(NEB)公司。
主要仪器:NanoDrop 2000(Thermo公司,美国);HiSeqTM 2500测序平台(Illumina公司,美国);Beckman Allgre 21R高速冷冻离心机(Beckman公司,美国);BC-subMIDI电泳仪(北京六一仪器厂);JY300C电泳槽(北京君意东方电泳设备有限公司);BioSens SC 810B凝胶成像仪(上海山富科学仪器有限公司);PTC-100PCR仪(MJ Research公司,美国);3730XL测序仪(ABI公司,美国)。
采用改良的CTAB法[4]提取所有样品DNA,利用1%琼脂糖电泳和NanoDrop 2000检测DNA的完整性和纯度。经纯度和完整性检验合格后将黄色底色单株和白色底色单株DNA分别等量混合成终浓度为40 ng/μL的黄色底色DNA混池和白色底色DNA混池,用于后续SLAF测序。
1.2.3文库构建、测序及SLAF标签开发
通过预实验发现已有的小兰屿蝴蝶兰(Phal.equestris)基因组和铁皮石斛(Dendrobiumcatenatum)基因组等兰科植物基因组对本研究采用的商品品种的参考性极低,因此采用无参考基因组的测序方法,以水稻基因组(Oryzasativa)为参考来评估实验建库的准确性,利用酶切反应预测软件SLAF_Predict 进行系统分析,根据其重复序列、GC含量和基因特点等确定采用RsaI+HaeIII双酶切对样本基因组DNA分别进行酶切。得到的酶切片段进行3′端加A处理、连接Dual-index测序接头、PCR扩增、纯化、混样和切胶选取目的片段,文库质检合格后用Illumina HiSeqTM2500测序平台进行测序。对测序得到的各样品的reads数据进行评估,通过reads间聚类的方法,在亲本和混池中开发SLAF标签。
1.2.4SNP_index关联分析
SNP_index是通过寻找混池之间基因型频率的显著差异,用Δ(SNP_index)统计从而进行标记关联分析的一种方法。Marker与性状关联度越强,Δ(SNP_index) 越接近于1。以大于99% Marker的Δ(SNP_index)的值作为阈值,大于该阈值的标记则为与性状显著关联的标记。计算方法如下:
SNP_index(ab)=Mab/(Pab+Mab)
(1)
SNP_index(aa)=Maa/(Paa+Maa)
(2)
Δ(SNP_index)= SNP_index(aa)-SNP_index(ab)
(3)
式中:Maa和Paa分别表示白色底色混池(aa)来源于母本和父本的深度;Mab和Pab分别表示黄色底色混池(ab)来源于母本和父本的深度。
1.2.5SNaPshot技术筛选SNP分子标记
根据关联分析获得的标记,参照Hommais等[15]的方法设计引物,在父母本和后代11个组群中每组挑选1株未在建库中使用的子代、2个白色底色种质资源和2个黄色底色种质资源验证并筛选底色相关SNP标记。预扩增采用10 μL反应体系:2 μL (20 ng/μL)样本基因组DNA、1 μL 10×Buffer I、0.8 μL dNTP、2 μL预扩增引物(F+R,5 μmol/L)、0.1 μL KAPATaqHotStart DNA 聚合酶 (5 U/μL)、4.1 μL ddH2O。反应程序:94 ℃预变性5 min;94 ℃变性30 s,退火(50~60 ℃)30 s,72 ℃延伸 30 s,共10个循环;94 ℃变性30 s,50 ℃退火30 s,72 ℃延伸30 s,共30个循环;72 ℃延伸10 min。取2 μL产物上2%琼脂糖凝胶电泳检测质量。每个样本的预扩增产物等比例混合后取4 μL,加入SAP酶1.33 μL,ExoI酶(20 U/μL)0.27 μL 混合成5.6 μL体系37 ℃酶切1 h,75 ℃变性20 min。取酶切后产物1.2 μL,加2 μL延伸引物混合物、0.5 μL ABI Mix、0.4 μL 10×Buffer I和0.9 μL ddH2O,96 ℃变性10 s、50 ℃退火5 s、60 ℃延伸30 s,共进行30个循环的延伸反应。取延伸反应产物6 μL 加入1 μL CIP酶,37 ℃ 1 h、75 ℃ 15 min进行纯化。96孔板中每孔加入分子量内标0.5 μL,甲酰胺8.5 μL,纯化后延伸产物1 μL,95 ℃ 变性3 min,于3730XL测序仪检测。
2 结果与分析
2.1 测序数据统计与评估
经Illumina HiSeq TM 2500测序平台进行测序,对父本、母本、白色底色混池和黄色底色混池测序数据的reads数量、Q30和GC含量等进行统计,共获得37.90 M reads数据,测序平均Q30为92.57%,平均GC含量为38.47%,具体结果见表1。测序质量值Q30是评估高通量测序单碱基错误率的重要指标,测序质量值越高对应的碱基测序错误率越低,本研究中4个样本的测序质量值Q30均在90%以上,说明测序碱基错误率低,所获数据合格。
第一,经由引导附近人们参与到营造林活动中的方式,有助于增加农村就业机会,促使广大农民可以经由投入劳务的方式获取相应的收入,对更好的维护和谐社会的稳定性具有积极意义。第二,经由项目建设,加大力度宣传以及应用多种现代化的经济林高产栽培技术等,有助于激发群众的种植发展热情,有助于推进产业发展进程。第三,对果树林实施有效的一次性种植操作,其稳产收获期超过二十年,有助于确保农民的收入稳定。
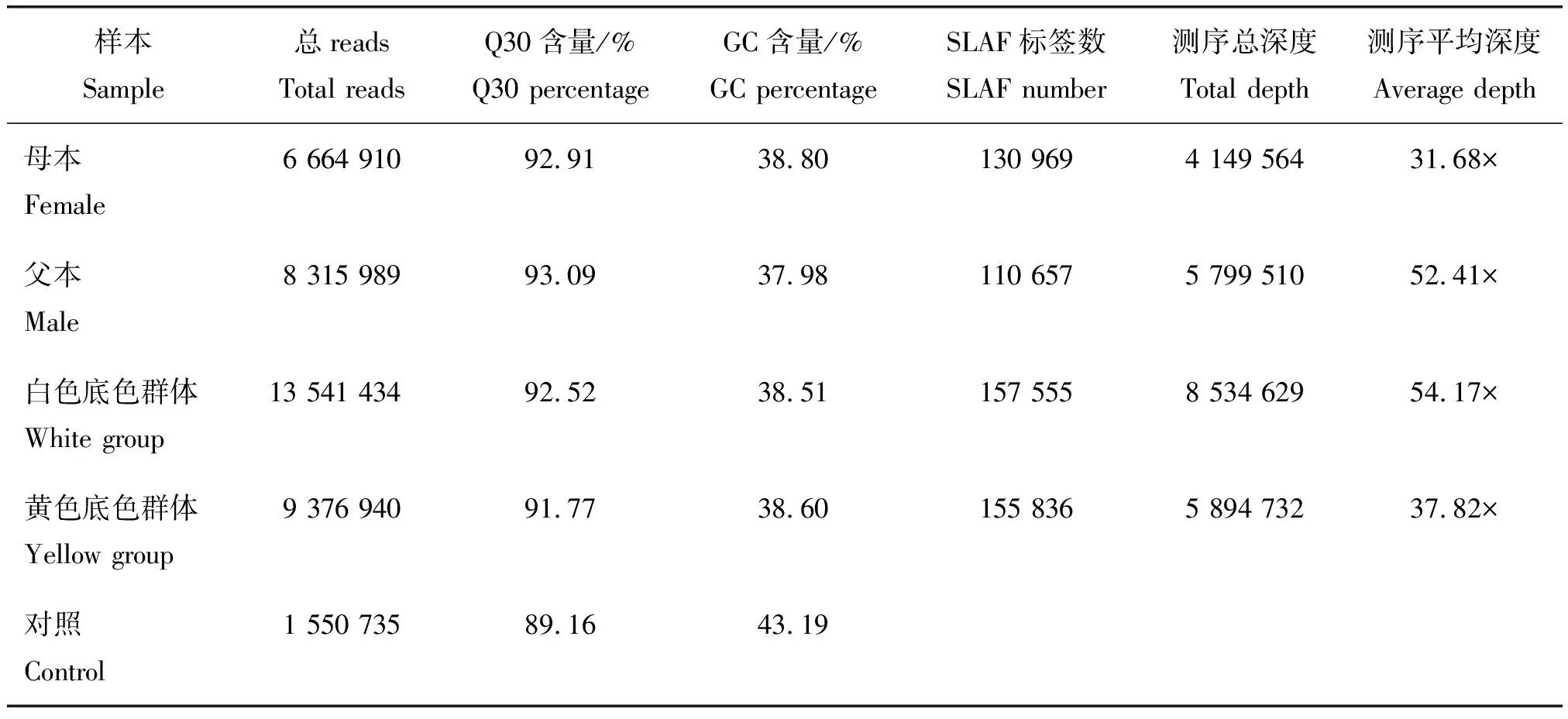
表1 样品测序数据评估和SLAF标签统计Table 1 Statistic results of sample sequencing data assessment and SLAF tag
2.2 SLAF标签和SNP标记的开发
剔除各组样本间的重复SLAF标签数,本研究在4组样本中共开发出164 874个不同的SLAF标签,SLAF标签亲本平均测序深度为42.05×,混池平均测序深度为46.00×。其中父本的测序深度为52.41×,母本的测序深度为31.68×,aa池的平均测序深度为54.17×,ab池的平均测序深度为37.82×。针对所有样本开发得到的SLAF标签,根据等位基因数和基因序列之间的差异进行多态性分析,共得到3种类型的SLAF标签:包含SNP/Indel多态性位点的多态型SLAF标签、没有多态性位点的非多态性SLAF标签和位于重复序列区的SLAF标签。其中多态性SLAF标签共有21 031个,多态性比例为12.76%。
2.3 关联分析
根据亲本基因型来源对得到的21 031个多态性SLAF标签,进行标记筛选。过滤亲本测序深度5×以下的标记,根据亲本的测序信息确定每个标记等位基因的亲本来源,选取一种基因型来源于父本,另一种基因型来源于母本的多态性SLAF标签共1 451 个进行后续的关联分析。通过Δ(SNP_index)的方法,以大于99% Marker的Δ(SNP_index)的值0.745 5作为阈值,筛选出15个与性状关联程度较显著的候选标记,含27个SNP位点(表2)。根据15个候选标记的扩增序列比对及注释分析,未能发现定位于功能基因上的SNP位点。
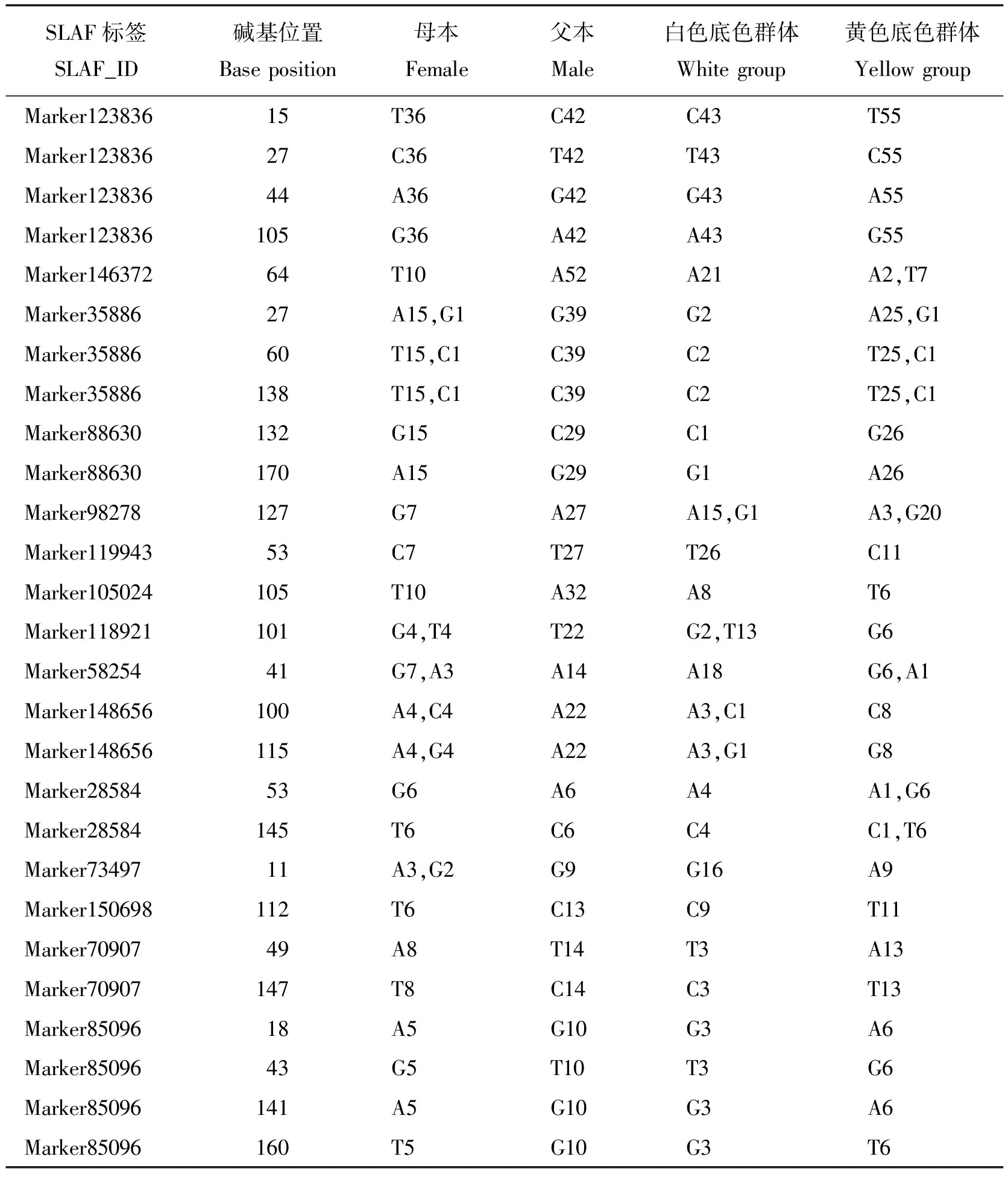
表2 与蝴蝶兰花底色性状关联的SNP标记Table 2 SNP markers associated with the flower ground color in Phal.
2.4 蝴蝶兰花部底色相关SNP分子标记的开发
根据关联分析获得的15个标记的序列信息和SNP所在位点,过于靠近序列5′端或3′端的SNP位点不能进行有效扩增和检测,设计出来自于14个标记片段上21个SNP位点的引物,通过对父母本、11个子代代表株和4个不同底色种质资源的基因组DNA为模板进行扩增和检测,筛选出2个引物Marker35886(SNP为扩增序列的第60位碱基)和Marker70907(SNP为扩增序列的第147位碱基)的SNP位点鉴定效果较好(引物序列信息见表3),在父母本中的SNP检测结果与SLAF测序结果一致,而在其他种质资源中的检测率分别达到 66.67% 和73.33%,2个引物联合使用的检测率达到93.33%,具体检测结果见表4。

表3 SNaPshot测序中Marker35886和Marker70907的相关引物序列信息Table 3 Primer sequences of Marker35886 and Marker70907 used in the SNaPshot
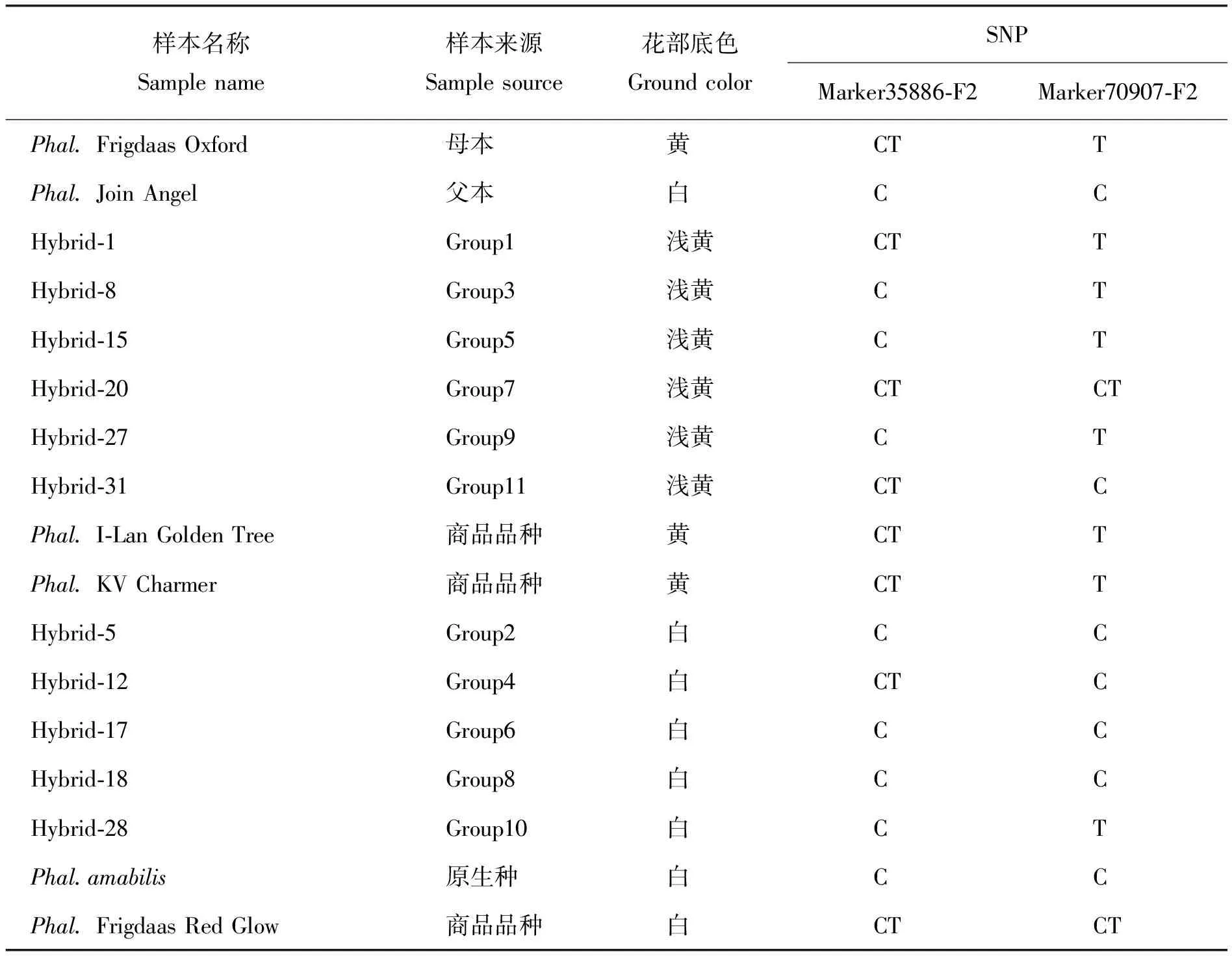
表4 Marker35886和Marker70907在不同种质资源中的检测结果Table 4 Detection results of Marker35886 and Marker70907 in different germplasm resources
3 讨 论
SLAF-BSA技术能够克服没有参考基因组和基因组复杂等问题,高通量地开发与复杂性状关联的SNP标记[7]。Ye等[11]通过SLAF-BSA技术筛选出2个与紫薇矮化性状高度关联的标签,组合检测效率可达93%;Li等[9]利用SLAF-BSA技术筛选到与红麻第一花节位紧密相关的一个SNP位点,检测效率达到91.2%;Wang等[10]通过SLAF-BSA技术筛选定位到12个基因与辣椒果实花青素的积累相关。本研究基于杂交F1代分离群体,利用SLAF-BSA技术在全基因组范围上筛选蝴蝶兰花瓣黄白底色相关SNP,并在未参与构建混池的后代和4个其他种质资源中进行验证,找到2个与蝴蝶兰黄白底色相关性较高的SNP位点,联合检测效率达93.33%,说明SLAF-BSA技术适用于兰科植物复杂性状相关标记筛选。
花色主要由花朵中花青素、类胡萝卜素及甜菜色素的含量及分布决定[16],并且还受到色素细胞的pH、金属离子、辅助色素[17]及花瓣表皮细胞形态[18]等因素的影响。目前,蝴蝶兰花朵中主要花色素的合成途径已较为清楚,关键结构基因和调控转录因子陆续被克隆和研究,但对整体调控机制的解析仍然很浅。研究发现,绝大部分花青素合成途径相关结构基因和转录因子的表达量在红色蝴蝶兰花瓣中都显著高于白色蝴蝶兰[19],UFGT基因与红花的形成高度相关[20],PeMyb2、PeMyb11和PeMyb12的不同表达比例会造成红色蝴蝶兰花瓣的不同颜色分布类型,沉默PeMyb2、PeMyb11和PeMyb12会分别导致整体红色、红色斑纹和红色脉络的消失[21]。黄色蝴蝶兰花瓣中F3’H、UF3GT、bHLH的表达量低于红色蝴蝶兰花瓣中的表达量,但PAL和PSY的表达量高于红色蝴蝶兰花瓣中的表达量[1]。紫色蝴蝶兰(Phal.schilieriana)花瓣中DFR的表达水平高于白色蝴蝶兰[22]。基于这些关键结构基因和调控转录因子的研究,Sudarsono等[23]
开发了一系列针对花色相关基因的SNP引物,并用于30份蝴蝶兰种质资源的聚类分析。但蝴蝶兰分子标记目前主要是用于种质资源的亲缘关系鉴定和品种鉴别上[24-25],很少有能够与性状关联的标记被开发出来[4,26]。在对兰科植物短距手参(Gymnadeniarhellicani)的研究中发现,SNP多态性导致一个R2R3-Myb转录因子提早终止表达,使得其调控的ANS基因表达量降低,花中红色花青素减少,从而出现了有别于野生型黑红色花的浅红色花和白色花[27]。但基于6个花色相关结构基因开发的SNP未能根据花色将不同颜色的蝴蝶兰资源区分开[23],表明兰科植物花色形成和调控机制极其复杂且具有特异性,花色相关SNP的筛选不能局限于个别已知相关基因,需扩大到全基因组范围内进行。
本研究利用SLAF-BSA技术在全基因组范围筛选到2个与蝴蝶兰花瓣黄白底色相关的SNP标记,联合检测效率达93.33%,可作为有效的分子标记位点在蝴蝶兰育种早期进行辅助选择。这样的检测效率在蝴蝶兰漫长的育种过程中,特别是旨在特定选择黄色底色或白色底色后代的育种计划中,是一个可接受的可靠水平。但是,本研究中鉴定出的是连锁标记而不是关联到了关键基因本身的标记,在实际育种过程中存在一定的局限性,且本研究采用的杂交群体母本及后代存在斑纹,尽管在构建2个不同底色混池的时候采用了斑纹对应分离的个体,尽量屏除了斑纹对后续建库分析的影响,但仍会在一定程度上影响筛选出的SNP位点与底色的相关性高低。另外,2个标记在杂交后代和其他种质资源中的扩增结果并不完全与SLAF测序结果一致,表明测序结果也存在一定技术层面的错误率。针对本研究存在的局限性,也基于蝴蝶兰花色这一复杂性状涉及众多基因和调控通路的现状,在单一杂交群体中筛选的少量相关SNP位点不能完全代表大量种质资源中的多态性,因此后续还需引入更多的研究手段对更多的种质资源进行分析和验证,进一步加强相关研究来完善蝴蝶兰花部底色关联分析。
4 结 论
通过对蝴蝶兰不同花瓣底色的亲本和杂交分离后代混池进行SLAF测序,共获得21 031个多态性SLAF标签,关联分析筛选出27个与黄白花瓣底色关联性较高的SNP位点,在未参与构建混池的后代和4个其他种质资源中进行验证,最终筛选得到2个联合检测效率达93.33%的SNP标记,表明SLAF-BSA分析方法可以用于兰科植物花色等复杂性状相关SNP位点的筛选。
