抗髓鞘少突胶质细胞糖蛋白G抗体相关疾病8例临床特点及康复结局研究
2021-11-08肖海兵杨清燕林慧婷崔玉真李黎娜叶晋豪卢镇泽曾文双褚晓凡
肖海兵, 杨清燕, 林慧婷, 崔玉真, 李 凌, 李黎娜, 叶晋豪, 卢镇泽, 曾文双, 黄 莹,2, 褚晓凡,2
中枢神经系统炎性脱髓鞘疾病是一类少见疾病的统称,该类疾病通常导致青壮年人群神经功能残疾,主要包括多发性硬化 (multiple sclerosis,MS)、视神经脊髓炎谱系疾病 (Neuromyelitis optic spectrum disorders,NMOSD)、抗髓鞘少突胶质细胞糖蛋白G抗体(anti-myelin oligodendrocyte glycoprotein-IgG,MOG-IgG)相关疾病 (MOG-IgG associated disorders,MOGAD)、急性播散性脑脊髓炎等。其中MOGAD是最近几年才被认识和描述的一种新疾病[1],在人民卫生出版社2015年第3版的八年制《神经病学》教材中,尚没有关于该病的章节。该病非常罕见,目前尚无其流行病学的确切数据,预估其患病率小于十万分之一[2]。然而,随着病例报道的积累,学者们逐渐认识到,MOGAD患者通常出现的视神经炎、脊髓炎、脑干脑炎和脑膜脑炎等症状,与MS、NMOSD的症状既有明显的重叠,又有显著的差异[3,4]。2018年,国际上刊出了两篇重要论文[5,6],一篇题为“MOG 脑脊髓炎诊断和抗体检测专家共识”,另一篇题为“MOG-IgG相关疾病的拟诊断标准”。这两篇论文的刊发,标志着国际主流学术界认可了MOGAD是一种独立的疾病谱。2020年,中国免疫学会神经免疫分会也基于中国经验刊发了《抗髓鞘少突胶质细胞糖蛋白免疫球蛋白G 抗体相关疾病诊断和治疗中国专家共识》(以下简称《MOGAD中国共识》),用于指导该病在我国的诊治[7]。然而,由于该病被认识的时间不长,目前报道的病例数也不多,有必要继续对该病的病例组进行报道,有利于提高临床及研究人员对该病的认识,也为后期该病的共识或指南修订提供依据。
1 资料与方法
1.1 研究对象 收集2016年1月至2020年12月在香港大学深圳医院成人神经科被诊断为MOGAD的所有病例(有1例患者由深圳市人民医院转诊而来,其影像学资料从该院获取)。作者根据中国免疫学会神经免疫分会2020年发布的《MOGAD中国共识》,对上述患者在我院的诊治档案进行重新回顾和分析,确保患者符合新版诊断标准,并进行门诊或电话访视。病例纳入标准:①符合2020年《MOGAD专家共识》的诊断标准;②曾在我院住院诊疗,且可通过面访或电话访视获取最新临床状态和预后信息;③采用全长人MOG作为靶抗原的细胞免疫荧光法检测血清MOG-IgG阳性。排除标准:①MOG-IgG低滴度阳性,且临床综合征不典型者或以其它诊断可更好解释者;②失访或拒绝回访者。本回顾性研究已得到香港大学深圳医院伦理委员会批准(No. 2015-12)。
1.2 研究方法 对符合上述标准的患者的资料进行收集和分析。包括:人口统计学(起病年龄和性别等)、临床特征(发病症状、有无复发,以及复发的症状)、影像学(主要是颅脑、脊髓及视神经的MRI)、血液和(或)脑脊液中枢神经脱髓鞘抗体、脑脊液及血液的常规生化检查、脑脊液寡克隆带、电生理测试(诱发电位)、眼科学检查(视力、视野、眼底测试等)、以及治疗方案。采用扩展的残疾状态量表(Expanded Disability Status Scale,EDSS)和改良的Rankin量表(modified Rankin scale,mRS)评分来评价患者的神经功能。因为视觉功能是该病最常受损的系统,故提取EDSS中视觉功能系统评分(visual function system scale,VFSS)对该项功能做单独分析。
所有患者的血清均被送至广州金域或广州欧蒙医学检验公司,采用细胞免疫荧光法检测水通道蛋白4抗体(Aquaporin 4 IgG antibody,AQP4-IgG)以及MOG-IgG。MOG-IgG检测时采用了全长人的MOG作为靶抗原。两例低滴度MOG-IgG阳性的患者血清亦被送往中山大学附属第三医院神经内科实验室接受了复核。
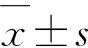
2 结 果
2.1 一般资料 最终纳入患者8例,男女比例1∶1。发病年龄20~56岁,中位年龄34岁。均为中国汉族人。起病至今病程最长108 m,最短者5 m,中位病程42 m;所有患者血清MOG-IgG均呈阳性,首次阳性结果中,1∶10低滴度阳性有3例(37.5%),1∶32高滴度阳性者有4例(50%),1∶320超高滴度阳性者1例(12.5%)(见表1)。
2.2 临床特征 8例患者中,首发症状表现为视神经炎的有4例(50%),均为单侧视神经炎;首发症状表现为脑膜脑炎或脑炎者2例(25%),表现为脊髓炎者2例(25%),其中1例病变在脊髓与延髓交界处,本文中将其归于脊髓炎。
对8例患者进行为期3 m至23 m随访,共有4例患者出现了复发,1例患者复发时表现为视神经炎合并显著脑膜炎,2例表现为单侧视神经炎,另外1例在首次发生脑炎后复发两次脑膜脑炎(或单纯脑炎)。首次MOG-IgG低滴度阳性(<1∶32)者复发率为33.3%,高滴度阳性者(≥1∶32)复发概率60%(见表1)。
2.3 脑脊液检查 八例患者有2例(25%)脑脊液白细胞数量正常,1例为视神经炎,另1例为短节段脊髓炎(病例2,颈髓与延髓交界处病灶)。其余6例(75%)脑脊液白细胞数量异常(>5×106/L),其脑脊液白细胞中位数为77×106/L (范围17~420×106/L)。其中表现为脑膜/脑炎或长节段脊髓炎的患者,脑脊液白细胞数量常>100×106/L,显著高于其它表现型者(见表1)。
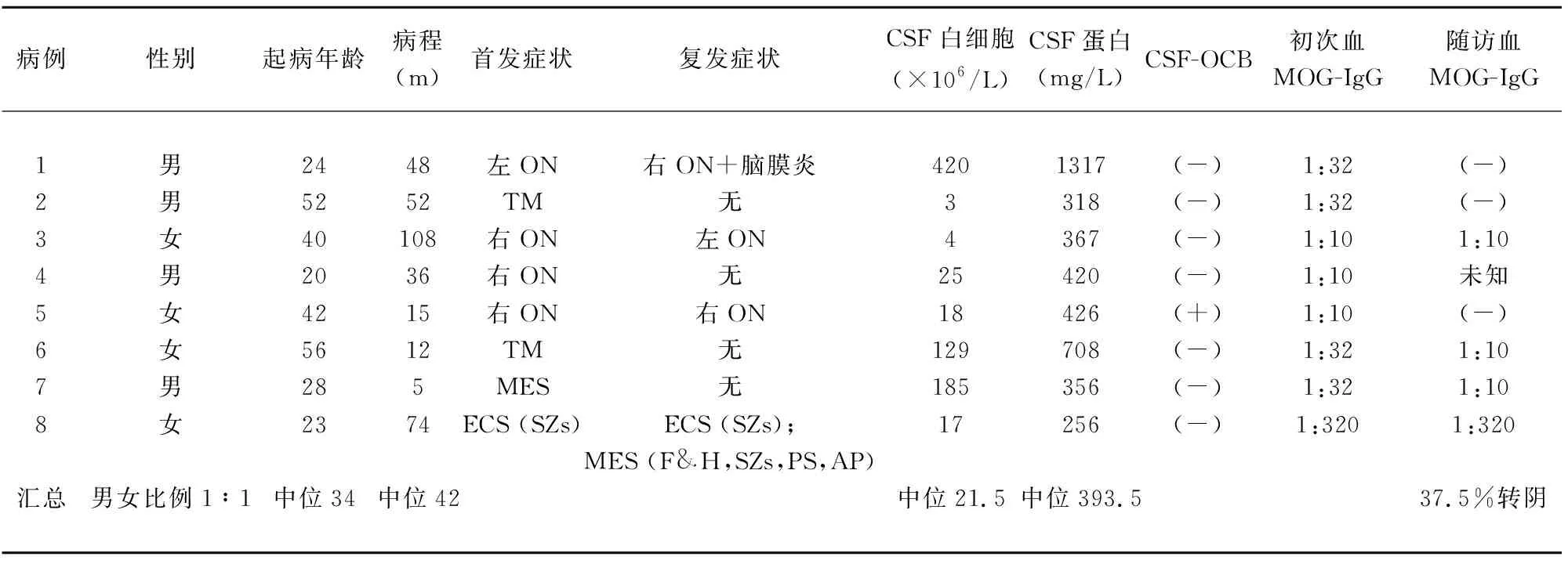
表1 MOGAD患者人口学、临床及检验情况
与细胞数异常率较高相比,患者的脑脊液蛋白含量的异常率不高。只有2例(25%)的患者被发现脑脊液蛋白增高。其中1例为病例1,表现为视神经炎合并脑膜炎,其脑脊液蛋白含量达到1317 mg/L;另1例为病例6,表现为长节段脊髓炎,其脑脊液蛋白量708 mg/L。
所有患者均接受了脑脊液寡克隆带分析,仅有1例患者(病例5)出现脑脊液寡克隆带阳性。该患者表现为复发性单侧视神经炎,颅脑MRI表现为类似多发性硬化的影像学特征。其余患者脑脊液寡克隆带均阴性。
2.4 影像学 在全部患者中,有2例(25%)患者眼眶增强磁共振、颅脑磁共振、脊髓MRI检查正常(均为视神经炎患者,病例1和3),其余6例(75%)出现不同的MRI异常。有1例视神经炎患者(病例4) 的颅脑MRI提示右侧间脑及左丘脑可见到高Flair信号的亚临床病灶伴局部强化(见图1,A-B),其眼眶MRI显示右侧视神经肿胀迂曲并强化,且伴随右眶内广泛T1增强信号(见图1,C、D)。另1例视神经炎患者(病例5)表现为皮质/近皮质、脑室旁、幕下的多发高T2/Flair亚临床病灶,类似于MS的影像学特征(见图1,E-H)。脊髓磁共振的表现则呈多样化,其中病例2表现为颈髓与延髓交界区短节段高T2/Flair病灶,呈偏心分布(图2,A、D)。病例6表现为分散的、长短不一的脊髓内高T2/Flair病灶,主要呈中心性分布(图2,E-H)。特征性的皮质信号异常主要出现在以脑炎/脑膜炎为首要临床症状的患者中,病例7表现为右侧额颞顶枕叶脑皮质的肿胀样高T2/Flair病灶(图3A)及同区域脑膜显著强化信号(图3B)。病例8在最后一次脑炎复发时出现左侧内外侧颞叶及枕叶的皮质肿胀,T2/Flair序列呈高信号(图3C),增强扫描时呈局部微弱的线状强化(图3D)。

A~D为1例右侧视神经炎患者(病例4):A:右侧间脑及左侧丘脑高Flair信号亚临床病灶;B:T1增强扫描可见右侧间脑不均匀片状强化;C:眼眶横断面MRI增强扫描可见右侧视神经与对侧比较明显呈肿胀迂曲且有强化;D:眼眶冠状位MRI增强扫描可见右侧视神经增粗、强化,右侧眶内组织与对侧相比也呈明显强化。E~H为另1例视神经炎患者(病例5)的颅脑MRI:Flair序列可见近皮质(E)、深部白质(F)、脑室旁(G)及幕下脑桥(H)广泛高信号脱髓鞘病灶

A-D为1例高颈段(脊延髓交界区)脊髓炎的颈椎MRI(病例2):A:T2序列可见高颈段脊延髓交界区高信号炎症病灶;B:为A图红色虚线处的横断面扫描,可见炎性病灶主要累及脊髓中心及后索;C: T1增强扫描见局部斑片状强化;D:T2压脂序列能更清晰地显示出病灶。E-H为另1例脊髓炎(病例6):E:T2扫描可见从C5-T3长节段脊髓内高信号(红实线箭头指示上界,红虚线箭头指示下界);F:为E图在红色虚线处横断面扫描,可见病灶在髓内呈中心分布;G:T2压脂序列更突出的显示出病灶信号;H:在T7节段还可见分散的小脊髓病变(蓝色箭头所示)

A-B为1例脑膜炎表现突出的脑膜脑炎患者(病例7):A:Flair序列各层面均显示右侧额顶枕颞皮质肿胀样高信号(如红箭头所指);B:T1增强扫描各层面均显示相应区域脑膜信号与左侧相比明显强化(如红箭头所指)。C-D为1例复发多次的脑炎患者(病例8):C:Flair序列左侧内外侧颞叶、枕叶均呈高信号肿胀(蓝箭头所指);D:T1增强扫描提示病灶强化不明显,仅内侧颞叶少许线状强化影
2.5 治疗及神经康复结局 各例患者在症状发作的急性高峰期均有程度不一的神经功能残疾。历经药物治疗和各种积极的康复措施,在最后随访期均获得了部分或者完全的康复(见表2)。7例患者(87.5%)在急性发作期接受了大剂量甲泼尼龙静脉注射冲击治疗(IVMP,方案为500~1000 mg×3~5 d,静脉滴注),然后逐渐减量(缓解期剂量为1 mg/kg*d,根据症状缓解程度,每1~2 w减量5~10 mg)。5例患者(62.5%)同时接受了大剂量免疫球蛋白注射治疗(IVIG,方案为0.4 g/Kg×5 d,静脉滴注),1例患者(12.5%)同时接受了血浆置换(隔天1.5L×5次)。有1例患者(病例1)在第一次发作时在外院诊断不明,未接受治疗,第二次发作表现为视神经炎合并脑膜炎,入住我院接受了经验性抗结核药物联合地塞米松(方案为20 mg/d,静脉静滴,后逐渐减量,临床症状亦得到部分康复。病例7在起病初期疑诊为病毒性脑炎同时接受了1 w的经验性阿昔洛韦抗病毒治疗。5例患者加用免疫抑制剂,均可控制病情发展。
有两例患者因为行走不稳(病例2)和肢体瘫痪(病例6),接受了物理治疗师的物理治疗(PT);有3例患者(病例6~8)同时接受了作业治疗师(OT)的康复治疗;有1例患者存在失语(病例8),同时接受了言语治疗师(ST)的康复训练(见表2)。
如表2所示,在发作急性期最高峰时,该病例组扩展残疾状态量表(EDSS)平均得分是(5±1.069)分,经治疗后在末次随访(末访)时下降至(2.19±1.689)分,差异有统计学意义。视觉功能状态评分(VFSS)在高峰时平均得分是(4.75±0.957)分,末访时下降至(3.25±0.957)分,虽然表现出下降趋势,但经分析显示差异无统计学意义。修订的Rankin残疾量表评分(mRS)在高峰时得分是(3.25±1.165)分,末访时其得分下降至(1.25±1.035),差异有统计学意义(表2)。上述结果显示,经过药物和康复治疗,该病例组总体神功能康复情况良好。

表2 MOGAD患者的症状、治疗以及神经康复结局
3 讨 论
MOGAD是最近数年被认识的一种罕见的、独立的中枢神经系统炎性脱髓鞘疾病。该疾病在成人主要累及青壮年人群,有明显的致残性和复发性,但同时又具备显著的可治疗性,因此对于该病进行充分地观察和报道是必要的。本文的MOGAD病例系列中,男女患病比率为1∶1,中位年龄34岁,无60岁以上发病者,这与文献报道基本一致[7]。
从临床分型而言,视神经炎仍然是该病最常见的表型,在本病例系列中,视神经炎占首发症状的50%,占全部临床症状的54%(见表1),与大多数文献报告是一致的。但个别研究中视神经炎占比高达90%,这可能与人种以及其研究人群包括小儿有关[8]。脑膜脑炎(或仅有脑膜炎/脑炎)是MOGAD常见的脑部症状,在本病例组中占全部发作的38.4%,这与同为中枢神经炎性脱髓鞘疾病的MS、NMOSD存在显著区别。我们还发现,表现为脑膜脑炎的患者由于脑脊液的炎性反应非常强烈(CSF细胞数通常>100×106/L),在就诊初期易被误诊为颅内感染,甚至被经验性地应用抗感染治疗。本文的第1号和7号病例就曾分别被予以抗结核[9]、抗病毒治疗(见表2)。其余患者表现为脊髓炎或者脑干脑炎。通常短节段的脊髓炎、单纯视神经炎的患者脑脊液的白细胞仅会轻度增高或正常(<50×106/L),而长节段脊髓炎或者脑膜/脑炎患者脑脊液白细胞增高很显著(>100×106/L)。其中病例5的脑脊液中还检测出寡克隆区带,提示MOGAD与MS的在病理机制上可能有一些相似之处。
在磁共振影像学表现上,MOGAD存在广泛多样的改变。如前所述,视神经炎作为该病最常见的临床表型,MRI通常阳性率不高,部分患者眼眶增强磁共振可表现为显著的受累侧视神经肿胀迂曲伴强化,且伴有眶内组织炎症强化(见图1,C-D),本文的发现与其他报道是类似的。在既往的研究中提出,皮质脑炎是该疾病的特征性改变,可以出现T2/Flair高信号的皮质肿胀改变。在我们的病例组中,脑膜/脑炎为临床症状的患者颅脑MRI出现显著的单侧的皮质肿胀,呈T2/Flair序列的高信号,可伴随脑膜显著或轻微强化(图3)。与文献报道的MOGAD脑炎MRI相比[10],本文病例系列的皮质影像学改变更加显著。另外,我们想重点提出的一个新观点是,MOGAD脑炎的脑皮质损害,往往是单侧性的,而非双侧性的,这在本文病例系列的病例7、病例8以及其他文献报道中均可证实[10~12]。这种单侧性病变,或许是区分于其它抗体所致的自身免疫性脑炎的影像学特征之一[13,14]。MOGAD的颅内病灶则较多样化,部分呈非特异性改变。本文病例组的病例5脑MRI表现为无症状的近皮质、深部白质、脑室旁及幕下脑桥多发Flair高信号脱髓鞘病灶,类似MS的典型MRI表现(见图1,E-H)。因此单纯从MRI上来鉴别MS和MOGAD是不够的,还需要相关抗体的检查以助诊断。MOGAD的脊髓MRI表现缺乏特异性,可表现为短节段偏心的T2/Flair高信号病灶,类似于MS的脊髓表现(见图2,A-D);亦可呈中心分布的长节段纵向延伸的横贯性脊髓炎,类似于NMOSD的脊髓炎改变(见图2,E-H)。
MOGAD的治疗分为急性期治疗和缓解期治疗两个方面。在急性期,通常以大剂量糖皮质激素冲击、或联用IVIG/血浆置换。缓解期通常采用泼尼松、硫唑嘌呤、吗替麦考酚酯口服,或者利妥昔单抗间断注射,这些治疗与MS不同,但与NMOSD的治疗是非常类似的[15]。在药物治疗的同时予以合适的康复手段(包括物理治疗、作业治疗、言语治疗等)会加速患者神经功能康复,这与其他中枢神经炎性脱髓鞘疾病的原理如出一辙[16]。在急性期,MOGAD有时会给患者带来显著的神经残疾(见表2),在我们的病例系列中,急性期最严重的残疾达到EDSS 7分、mRS 5分。欣慰的是,经过药物和康复手段的联合治疗,该病例系列的EDSS评分从急性期平均(5.00±1.069)分恢复到末次随访时(2.19±1.689)分,mRS也从高峰期(3.25±1.165)下降到末访时(1.25±1.035),上述差异均有统计学意义(见表2)。这提示了该病经过规范治疗,具备良好的康复结局。视觉功能系统评分(VFSS)的恢复没有统计学差异,这提示视神经损伤后的代偿能力较差,同时也提示在诊断该病之后,更需要积极治疗,避免视神经炎再发。
有较多的文献指出[4,7],血清MOG抗体的滴度是否下降或转阴,与疾病的活动性有关,在我们的病例组中,第8号病例持续存在1:320高滴度的MOG抗体,临床上也反复的发生脑膜/脑炎。这提示我们在治疗随访中,需要监测MOG-IgG滴度,如果滴度显著减低或转阴,可以考虑减少或停止免疫药物,以避免不必要的药物副作用。
总之,MOGAD是一种罕见的中枢神经脱髓鞘疾病。虽然近年来与MOGAD相关的临床与基础研究获得了快速发展,亦有国内国际专家共识发表,但很多神经科同行尤其是基层医生对该病还很陌生。我们的病例数虽然不多,但随访时间长、影像资料及治疗预后数据较为完整,相信会给同行们带来有关MOGAD更丰富的认识,从而提升MOGAD的防治工作。
