Selective annulation of benzamides with internal alkynes catalyzed by an electron-deficient rhodium catalyst
2021-11-06PingZhngWenjuChngHongyunJioYnshngKngWenxunZhoPeipeiCuiYongLingWeiYinSunYiLu
Ping Zhng,Wenju Chng,Hongyun Jio,Ynshng Kng,Wenxun Zho,Peipei Cui,Yong Ling,Wei-Yin Sun,Yi Lu,*
a Coordination Chemistry Institute, State Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Nanjing National Laboratory of Microstructures, Collaborative Innovation Center of Advanced Microstructures, Nanjing University, Nanjing 210023, China
b State Key Laboratory of Coordination Chemistry, Jiangsu Key Laboratory of Advanced Organic Materials, School of Chemistry and Chemical Engineering,Nanjing University, Nanjing 210023, China
ABSTRACT An electron-deficient[CpERhCl2]2 catalyzed annulation of N-pentafluorophenylbenzamides with internal alkynes was successfully established under mild reaction conditions, with the assistance of Lewis acid silver salt.Particularly, electron-deficient benzamide substrates were smoothly transformed into the desired products in this catalytic system.The catalytic system showed a broad tolerance for different substituents on the aromatic rings or aryl, alkyl-substituted alkynes.
Keywords:Electron-deficient rhodium C--H activation Annulation Benzamide Alkyne
The synthesis of small molecules through rhodium-catalyzed C--H activation has been greatly developed in the past few years[1-4].Generally, a commercially available [Cp*RhCl2]2(Cp* =pentamethylcyclopentadienyl)and its cationic complex are nearly universally considered to be privileged catalysts for these transformations[5-15].It attributes the success to the strongπ-electron donating ability of the Cp* ligand to obtain labile intermediates such as cationic or high-valent metal species [16,17].Recent studies have showed that some sterically or electronically tuned Cp ligands can dramatically induce significant changes in reactivity,as well as chemo-, regio- and diastereo-selectivity for different reactions.Satoh and Miura groups reported that the addition of phenyl-substituted Cp used as ligands improved the reactivity and changed the chemo-selectivity in rhodium-catalyzed C--H bond activation [18-20].With the development of the steric and electronic manipulation of Cp ligands, [CpXRh] catalysts have been used in rhodium-catalyzed C--H bond functionalization[21-24].Tanaka group demonstrated that electron-deficient CpEligand could improve reaction reactivity in rhodium catalytic system(Scheme 1a) [22].Chang [16] and co-workers reported that tuning the electronic property of Cp ligand could also change the chemo-selectivity (Scheme 1b).At the same time, enantioselective Rh-catalyzed C--H functionalization reactions promoted by the introduction of chiral Cp ligands were disclosed by Cramer[25-30], Li [31-34], You [35,36] and others [37-40].

Scheme 1.Cyclopentadienyl ligands in Rh(III)-catalyzed C--H activation reactions.
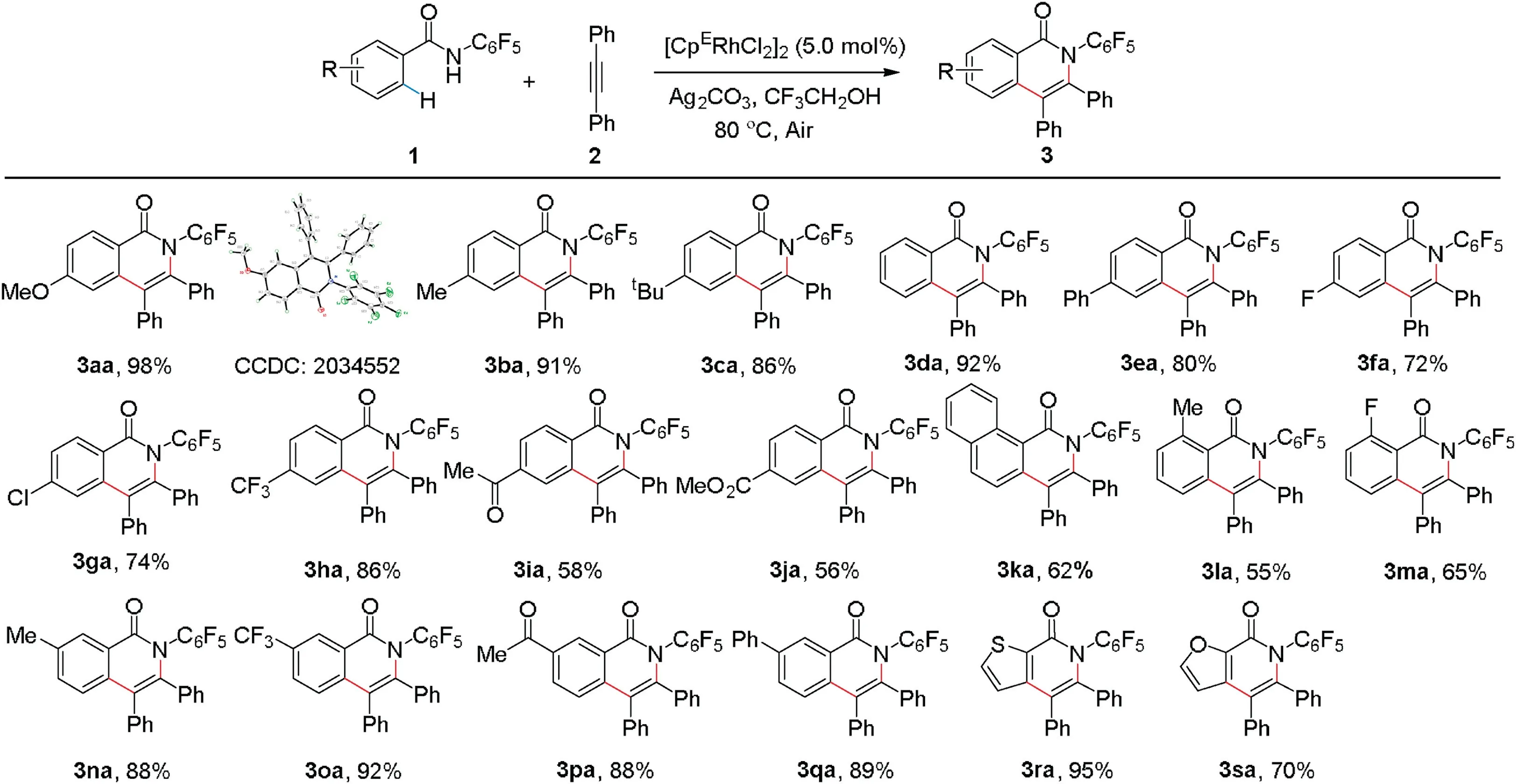
Scheme 2.Scope of benzamides.
The nitrogen-containing heterocycles in biologically active molecules have attracted much attention for their synthesis and functionalization in rhodium catalytic systems [6-11,41-46].Recently, the development in Rh chemistry has demonstrated that CpERhIIIcomplex bearing two ester groups could successfully realize the construction of heterocycles through C--H activation[16,22-24,47-50].Based on these progresses and our previous work [51-55], we developed an oxidative selective annulation of benzamides with alkynes catalyzed by the electron-deficient rhodium complex CpERhIIIin the air under mild conditions, with the assistance of Lewis acid silver salt.
On the basis of our work[55],we tried to employ an electrondeficient[CpERhCl2]2as a catalyst in place of[Cp*RhCl2]2in CH3CN solvent at 100°C (Table 1, entry 2).To our delight, it afforded a single isomer 3aa in 28% yield.Subsequently, we were pleased to find that the yield was improved dramatically when CF3CH2OH was applied as solvent (entry 7).Investigation of oxidants proved that Ag2CO3was most effective(entries 8-11).Generally speaking,[CpERhCl2]2was not suitable for the C--H bond functionalization of electron-deficient substrates according to the literature [23].However, in this work, electron-deficient benzamides were smoothly transformed into the desired products with the catalyst[CpERhCl2]2, while catalysts with Cp* and Cp*Cydecreased the chemo-selectivity(entries 14 and 15).It is probably because of the electronic effect of N-pentafluorophenyl, which enhances the acidity of the hydrogen atom on the N--H bond.With the assistance of carbonate anion from Ag2CO3, the nitrogen anion is more readily coordinated to the electron-deficient rhodium(III)complex.Moreover, tests of several substrates with different directing groups proves the importance of electronic effects as well(Table S5 in Supporting information).Compared to the substrates with electron-donating N-methyl and -benzyl groups, thesubstrate with N-methoxy group is relatively easier to be deprotonated by carbonate ion in this catalytic system.The steric and electronic properties of substrates with N-phenyl group are similar to the one with N-methoxy group.In the presence of strongly electron deficient N-pentafluorophenyl group, hydrogen proton is more easily removed with the assistance of carbonate ion,providing the best result for this transformation.
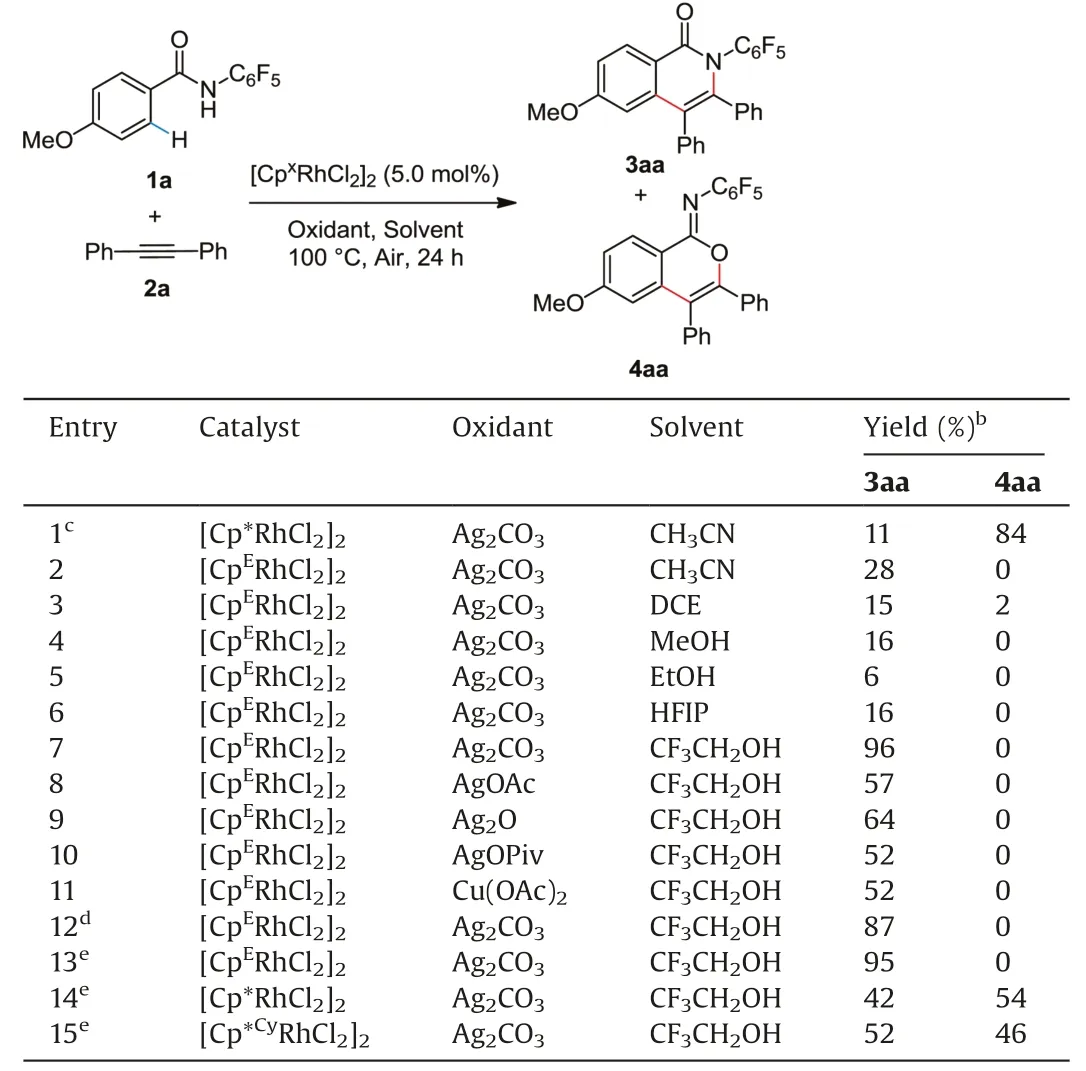
Table 1 Screening of reaction conditions for 3aa.a
With the optimized reaction conditions in hand, we explored the scope of this approach for products 3 by employing different substituted benzamides under the optimal reaction conditions(Scheme 2).Various substituted benzamides 1 were treated with 2a to give the isoquinolone derivatives in moderate to excellent yields.Both electron-donating(3aa-3ea)and electron-withdrawing (3fa-3ka) groups at the para position on the substrates were well tolerated.Substrate 1 with methoxy-group at the para position showed the highest reactivity.The structure of 3aa was confirmed by single-crystal X-ray analysis (Section 2.5 in Supporting information).2-Fluorobenzamide afforded 3ma in moderate yield (65%), whereas 2-methylbenzamide showed a slightly decreased efficiency (3la).It is noteworthy that benzamides possessing different substituents, such as methyl, trifluoromethyl,acetyl,methoxycarbonyl,and phenyl groups at the metaposition could lead to the formation of a single regioisomer(3na-3qa), attributing to the steric effects [56].Furthermore, heteroaromatic benzamides also turned out to be compatible(3ra,3sa).
As shown in Scheme 3, the catalytic system was not only available to diphenylacetylene (2a) but also suitable for aryl- or alkyl-substituted alkynes.Symmetrical diaryl or dialkyl alkynes led to the products in moderate to good yields (3ab-3ag).Intriguingly, unsymmetric 1-phenyl-3,3-diethoxy-1-propyne was applied as a coupling partner,and it proceeded smoothly to afford 3ah (CCDC: 2050115) in 84% yield with high regioselectivity(Section 2.5 in Supporting information).However,when 1-phenyl-1-pentyne was applied,a mixture of regioisomers 3ai and 3ai'was produced.
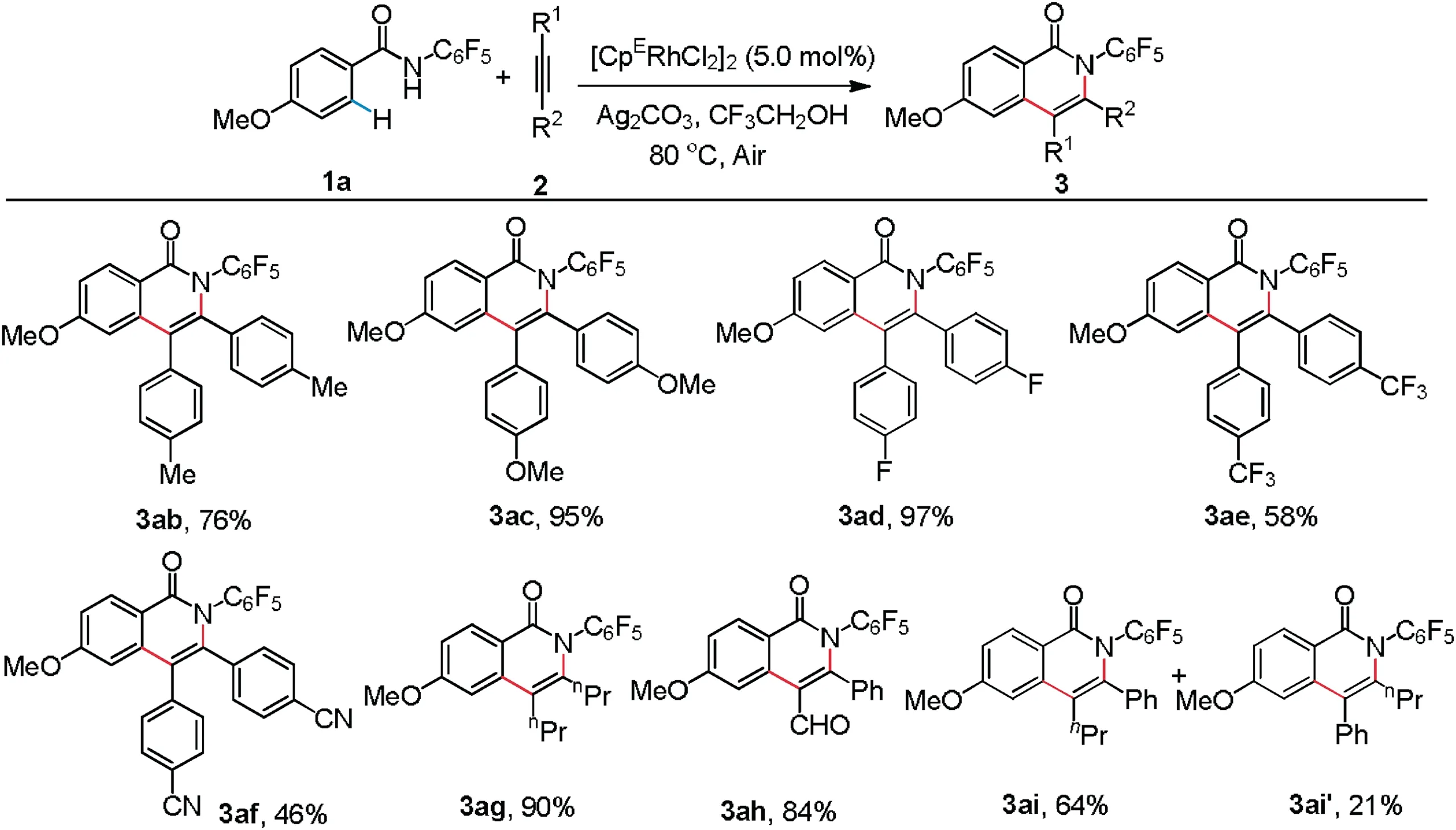
Scheme 3.Scope of alkynes.For compound 3ah,1-phenyl-3,3-diethoxy-1-propyne was used.
To understand the chemo-selectivity of this reaction, we performed a series of density functional theory(DFT)calculations using 1,2-diphenylacetylene (2a) and amide 1d as the substrates[57-61].The calculations demonstrated that the reaction proceeded through a C-H activation, alkyne insertion, and metallallylcarbenoid annulation (Scheme 4, for computational details,see Section 2.6.2 in Supporting information) [62].The chemoselectivity was determined in the C--H activation step via transition states TS-N-Ag-1 (17.6 kcal/mol) and TS-O-Ag-1(22.2 kcal/mol).Therefore, the N-directed pathway was favored over O-directed pathway by 4.6 kcal/mol, in agreement with that only N-cyclization product 3da is observed in the presence of CpERh catalyst.In the N-directed pathway,the C--H activation step(via TS-N-Ag-1) is 1.7 kcal/mol higher than the alkyne insertion(via TS-N-Ag-2)indicating an ambiguous kinetic isotope effect.The kinetic isotope experiments were conducted with 1d and 1d-d5under standard reaction conditions (parallel experiment:kH/kD=1.50 and intermolecular: kH/kD=1.55), which were consistent with DFT calculations.
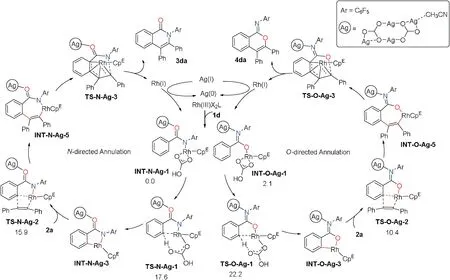
Scheme 4.Proposed mechanism.Numbers associated with molecules are DFT-computed relative Gibbs free energy (kcal/mol) in CH3CN.
In summary, we have successfully developed a selective Nannulation of benzamides with internal alkynes under mild reaction conditions with an electron-deficient catalyst[CpERhCl2]2.The electronic effect of ligand played a vital role in controlling the chemo-selectivity of this transformation.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We gratefully acknowledge the National Natural Science Foundation of China (Nos.21671097 and 21573106) and the Fundamental Research Funds for the Central Universities for financial support.
Appendix A.Supplementary data
Supplementary data associated with this article can be found,in the online version, at https://doi.org/10.1016/j.cclet.2021.01.024.
杂志排行

Chinese Chemical Letters的其它文章
- Molecular recognition triggered aptazyme cascade for ultrasensitive detection of exosomes in clinical serum samples
- Synthesis of sponge-like TiO2 with surface-phase junctions for enhanced visible-light photocatalytic performance
- Zn-based metal organic framework derivative with uniform metal sites and hierarchical pores for efficient adsorption of formaldehyde
- High active amorphous Co(OH)2 nanocages as peroxymonosulfate activator for boosting acetaminophen degradation and DFT calculation
- Effects of the molluscicide candidate PPU06 on alkaline phosphatase in the golden apple snails determined using a near-infrared fluorescent probe
- A lysosomal polarity-specific two-photon fluorescent probe for visualization of autophagy