超高效液相色谱-串联质谱法测定菊花15种吡咯里西啶生物碱毒素
2021-11-05章豪吴银良朱勇赵健陈国
章豪,吴银良,朱勇,赵健,陈国
(宁波市农业科学研究院 农业农村部农产品质量安全风险评估实验室(宁波),浙江 宁波 315040)
吡咯里西啶生物碱(PAs)是植物抵御昆虫及食草动物等侵害的一类化学物质,广泛分布在植物中[1]。目前,已在菊科的植物中检测到PAs[2]。联合国粮农组织和世界卫生组织(WHO)认为,PAs是草药产品与食品中最广泛和最严重的掺杂性与内源性的毒性成分[3]。PAs的毒性与结构密切相关,可形成烯丙酯结构的PAs具有肝脏毒性,会造成肝硬化、肝细胞坏死等症状[4]。欧盟于2019年实施PAs限量规定,成人每天摄入量不超过0.35 μg,儿童不超过0.14 μg[5]。WTO贸易技术壁垒委员会曾因我国出口的某种药材中PAs含量过高而发布通告禁令;欧盟食品和饲料类快速预警系统也曾因我国出口至欧盟的草本茶原料中PAs含量过高发布警告。我国法定药材标准中已收载了菊花等多种含有PAs的药材,但对PAs在菊花中的含量及种类等缺乏相关的数据。目前,我国尚未建立高效的检测方法,因此,还未能准确评估其在菊花中的含量水平,亟须对我国常见菊花中PAs的检测方法进行研究,进一步通过摸底检测明确菊花中PAs含量数据,以期厘清我国菊花食品安全风险,确定关键控制点,形成管控措施,应对潜在的国际贸易壁垒风险。
目前,针对PAs残留采用的分析方法主要为液相色谱-串联质谱法(LC-MS/MS)[6-9]、液相色谱法(LC)[10-12]、气相色谱-质谱法(GC-MS)[13-15]、气相色谱法(GC)[16-19]、分光光度法[20]、核磁共振法[21]和免疫学方法[22]。但使用分光光度法、核磁共振法和免疫学方法时存在基体干扰大的问题,更多是用来筛选,难以对多种PAs毒素进行分析。而GC和GC-MS分析PAs存在过程烦琐、耗时长、稳定性差、检出限高等缺陷,PAs分析使用 GC及GC-MS 较少,LC或LC-MS/MS是PAs主要分析手段。超高效液相色谱-串联质谱法(UPLC-MS/MS)在 PAs 分析中选择性好、灵敏度高,且适用于同时检测多种 PAs,因而广泛用于农产品或食品中PAs 的定性定量分析[9]。
PAs是含有特征性碱性氮的还原性生物碱,通常使用半极性或极性有机溶剂或在酸化水条件下萃取,其氧化形式(PANOs)属于极性分子,容易被极性溶剂(例如甲醇)或稀酸水溶液萃取[6]。PAs前处理技术主要包括液液萃取(LLE)法、固相萃取(SPE)法、QuEChERS 法,以及发展的新型技术如分散液相微萃取(DLLME)、固相支持液/液萃取(SLE)[6-19]。其中,SPE是常用净化PAs的前处理方法。常用固相萃取柱有C18和C8柱[8-11]。它们存在共提物多的缺点,在色谱分析中存在大的背景干扰,会对定量分析的准确性以及灵敏度产生影响[7]。对于生物碱的净化,阳离子交换柱是一种更为有效的纯化方法,可以有效地去除弱极性与离子型极性干扰物质,对PAs及其氮氧化物均有良好的纯化效果,并且不受其他共流出物的干扰[8]。
我们通过优化仪器条件和样品前处理方法,建立了基于Oasis MCX固相萃取柱能同时对菊花15种PAs毒素残留进行UPLC-MS/MS分析的方法。该方法具有精密度和准确度高、检测耗时短的特点,能同时对菊花样品多种PAs毒素进行测定。
1 材料与方法
1.1 材料
UPLC XevoTMTQ-S micro 超高效液相色谱-串联质谱仪,配有电喷雾离子源(ESI)及MassLynxV4.1数据处理系统(美国Waters公司);低温高速离心机(德国Sigma公司);Vortex Genie 3 漩涡振荡器(德国IKA公司)。
PAs标准品(天芥菜碱、天芥菜碱-N-氧化物、倒千里光碱、倒千里光碱-N-氧化物、千里光宁碱、千里光宁碱-N-氧化物、千里光碱、千里光碱-N-氧化物、促黑激素、促黑激素-N-氧化物、千里光菲灵碱、千里光菲灵碱-N-氧化物、欧天芥菜碱、欧天芥菜碱-N-氧化物和克氏千里光碱)纯度大于95%(德国PhytoLab GmbH & Co. KG公司);甲酸(色谱纯,美国Tedia公司);乙腈、甲醇(色谱纯,德国Merck公司);Oasis MCX固相萃取小柱(200 mg/6 mL,美国Waters公司);实验用水为Milli-Q超纯水。
1.2 方法
1.2.1 标准溶液配制
分别准确称取适量(精确至0.1 mg)15种PAs标准品,用甲醇溶解,配制质量浓度为1 g·L-1的标准储备液,于-20 ℃ 保存;分别准确吸取1 mL标准储备液,置于100 mL容量瓶中,用甲醇稀释,配制质量浓度为10 mg·L-1的混合标准储备液,于-20 ℃保存。
基质匹配标准溶液。制备菊花空白样品溶液,样品处理方法采用1.2.2节,通过稀释混合标准储备液配制基质匹配标准溶液。
1.2.2 样品处理
称取5.00 g(精确至0.01 g)菊花样品置于50 mL离心管中,加入10 mL 0.1 mol·L-1硫酸水溶液,10 000 r·min-1均质1 min,超声提取15 min;以5 000 r·min-1速度离心10 min,将上清液收集,残渣再用10 mL 0.1 mol·L-1硫酸水溶液提取1次,合并提取液。分别采用5 mL甲醇与5 mL去离子水活化Oasis MCX固相萃取小柱,移取提取液至固相萃取小柱,待液面到达柱床表面时用5 mL水淋洗,再用1%甲酸水溶液淋洗,最后用5 mL 0.5%氨水甲醇溶液洗脱,洗脱液在40 ℃下氮气吹干;用1 mL 0.1%(V/V)甲酸水-甲醇(90∶10,V/V)溶液复溶,过0.22 μm滤膜,供UPLC-MS/MS分析。
1.2.3 色谱条件
色谱柱:Acquity UPLC BEH C18(1.7 μm,100 mm×2.1 mm);流动相:A相为0.1%(V/V)甲酸水溶液,B相为甲醇;梯度洗脱程序:0~1.0 min,10%B;1.0~7.0 min,10%B~90%B;7.0~8.0 min,90%B;8.0~8.1 min,90%B~10%B;8.1~10.0 min,10%B;流速0.30 mL·min-1;进样量为10 μL。
1.2.4 质谱条件
多反应离子监测(MRM)模式;ESI源正离子模式电离;锥孔气流速:50 L·h-1;脱溶剂气流速:1 000 L·h-1;脱溶剂气温度:500 ℃;离子源温度:150 ℃;毛细管电压:2.0 kV;萃取锥孔电压:20 V;RF透镜电压:0.5 V;15种PAs毒素的母离子、子离子及其锥孔电压和碰撞能量见表1。
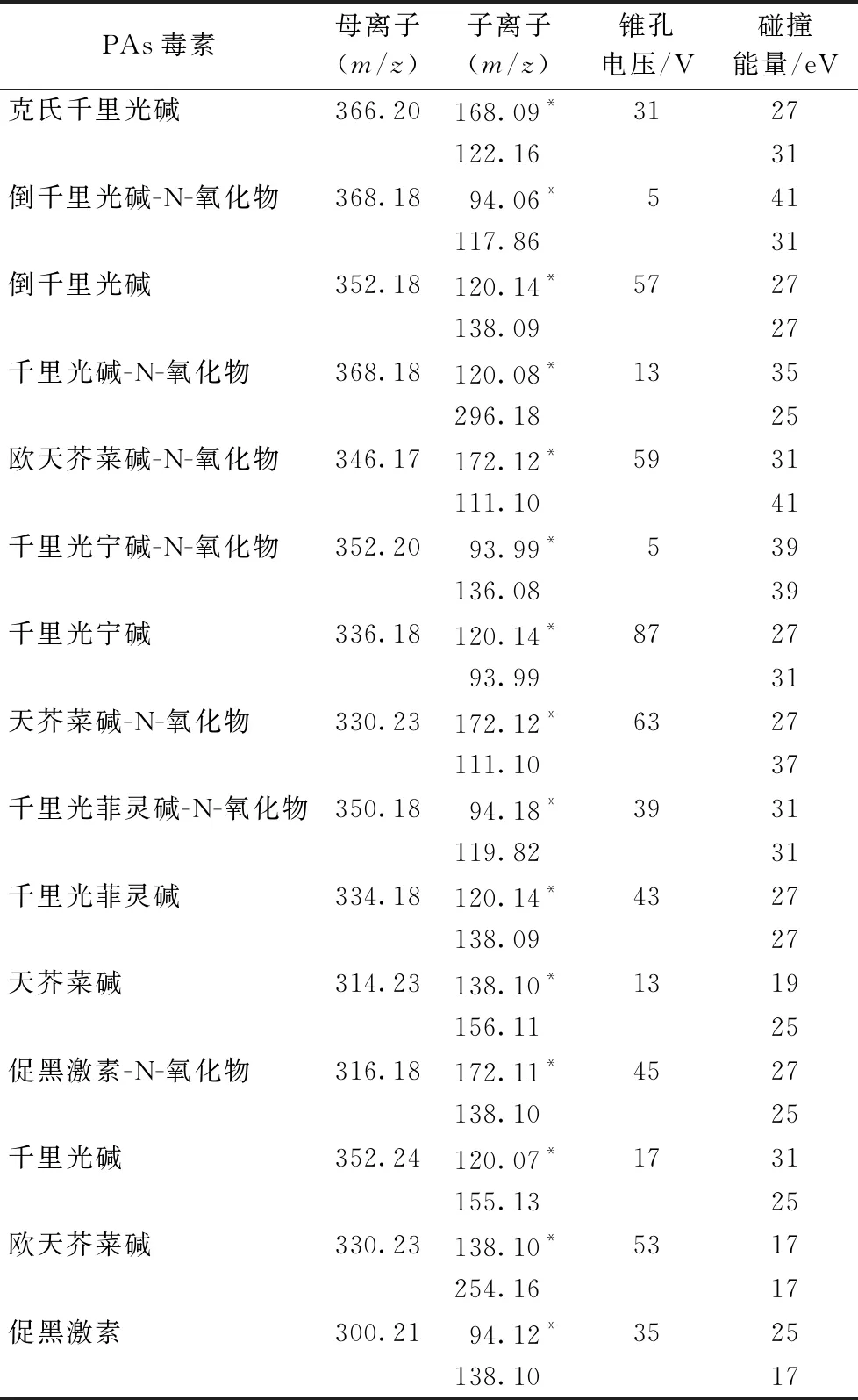
表1 15种PAs毒素的母离子、子离子及其锥孔电压和碰撞能量
2 结果与分析
2.1 仪器条件优化
在使用液相色谱-串联质谱法中多采用ESI源正离子电离模式来测定PAs毒素残留[6-9]。采用甲醇分别配制1.0 mg·L-115种PAs毒素标准溶液,通过Intellistart软件分别摸索15种PAs毒素的母离子、子离子及其锥孔电压和碰撞能量(表1),15种PAs毒素在ESI源正离子电离模式下均可获得较好的响应。
考查乙腈和甲醇作为色谱流动相的有机相时PAs毒素的分离效果,发现当采用乙腈作为有机相时,千里光碱、千里光菲灵碱-N-氧化物和倒千里光碱-N-氧化物的响应较差,而采用甲醇作为有机相时,15种PAs毒素的响应均较好,因此,确定甲醇为有机相。
2.2 前处理条件优化
PAs的提取,溶剂一般选用极性较大的溶液如甲醇[6-7]或水[8-9]等,加入少量的酸,可提高PAs的水溶性[14-17]。因此,本研究使用甲醇、水和0.05 mol·L-1硫酸水溶液3种溶剂对菊花样品在10 μg·kg-1加标水平下进行提取。发现不加酸提取时,采用水提取所有PAs毒素的回收率均低于采用甲醇提取的回收率;而加酸提取时,采用0.05 mol·L-1硫酸水提取所有PAs毒素的回收率均高于采用甲醇提取的回收率,因此,确定以0.05 mol·L-1硫酸水溶液进行提取。这可能是因为PAs毒素以离子形式存在于酸性环境中,显著增强其水溶性,导致酸水的提取效率比甲醇高。
2.3 基质效应、标准曲线、检出限和定量限
在考查菊花基质中15种PAs毒素的基质效应时我们采用基质匹配标准曲线的斜率/溶剂标准曲线的斜率×100%的计算方法。结果越接近100%,表明基质效应越小。当结果小于100%时表明存在基质抑制效应,大于100%时表明存在基质增强效应。表2显示,15种PAs毒素均存在一定的基质抑制效应或是增强效应。为了消除基质效应所带来的影响,我们采用基质匹配标准曲线来对目标化合物进行测定。采用菊花空白基质液配制1~1 000 μg·L-1横跨3个数量级的系列基质标准工作溶液(其中千里光碱、千里光菲灵碱-N-氧化物和倒千里光碱-N-氧化物为5~1 000 μg·L-1)进行UPLC-MS/MS测定分析,通过峰面积对浓度作标准曲线,从而得到线性回归方程。15种PAs毒素在相应浓度范围内相关系数均大于0.999,线性关系良好。通过3倍信噪比(S/N=3)进行估算,15种PAs毒素的检出限为0.5~9.0 μg·kg-1;通过10倍信噪比(S/N=10)进行估算,15种PAs毒素的定量下限为1.7~30.0 μg·kg-1。

表2 15种PAs毒素在杭白菊基质中的标准曲线、相关系数、检出限、定量限和基质效应
2.4 回收率与精密度
对杭白菊进行添加回收试验,并计算相应的相对标准偏差。杭白菊空白样品中PAs毒素的加标MRM色谱图如图1所示。杭白菊中千里光碱、千里光菲灵碱-N-氧化物和倒千里光碱-N-氧化物的添加量为30 μg·kg-1,其余12种PAs毒素的添加量为6 μg·kg-1,每批次同一浓度5个平行样品,重复测定3次。

图1 杭白菊加标样品的MRM色谱
表3显示,15种PAs毒素的加标回收率为82.6%~94.2%,相对标准偏差为2.3%~5.0%。
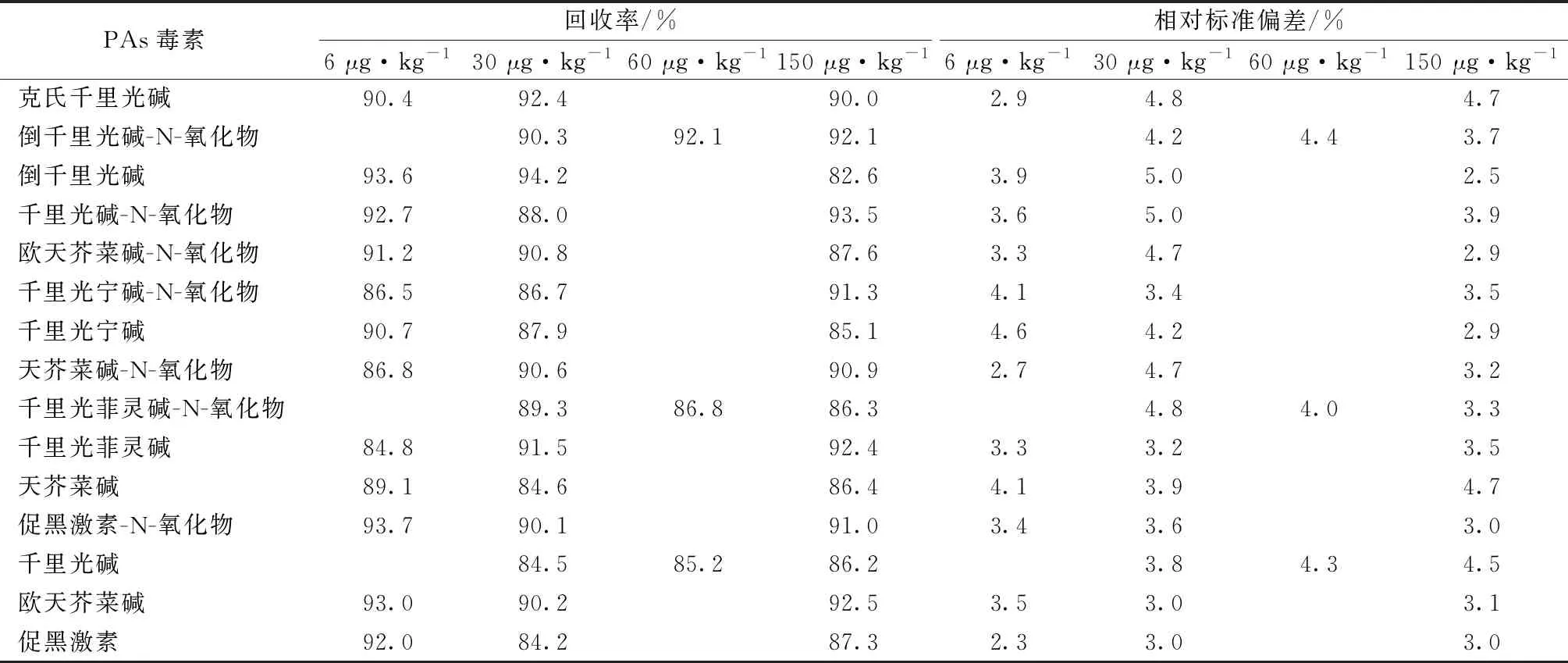
表3 15种PAs毒素在杭白菊中的回收率和相对标准偏差(n=5)
结果表明,方法对于15种PAs毒素在杭白菊中的残留分析均具有较好的精密度和准确度。
3 小结
建立了同时对菊花15种PAs毒素残留进行测定的UPLC-MS/MS方法,采用硫酸溶液提取菊花样品中PAs毒素,离心后将上清液过Oasis MCX固相萃取小柱净化,氮气吹干后复溶,采用UPLC-MS/MS进行分析。15种PAs毒素的添加回收率为82.6%~94.2%,相对标准偏差为2.3%~5.0%;检出限为0.5~9.0 μg·kg-1,定量下限为1.7~30.0 μg·kg-1。该方法检测耗时短,具有较高的精密度和准确度,具备同时测定菊花样品中多种PAs毒素的能力。
