基于转录组数据的三雌蕊小麦中SNP/InDel位点的挖掘
2021-11-01国钰环YAMAMOTONaoki彭正松廖明莉魏淑红吴一超杨在君
国钰环,YAMAMOTO Naoki,彭正松,廖明莉,魏淑红,吴一超,杨在君
(1.西华师范大学 生命科学学院,西南野生动植物资源保护教育部重点实验室,四川 南充 637009;2.西昌学院 农业科学学院,四川 西昌 615013)
小麦(TriticumaestivumL.)是全球最重要的谷类作物之一,为人类提供了大约20%的食物来源,在农业生产中占有极其重要的地位。全世界小麦的总种植面积大约为2.2亿 hm2,其中我国的种植面积为2 400万 hm2[1-2],从种植面积来看,我国是世界上最大的小麦生产国,但就其产量而言,我国的平均产量仅有5 481 kg/hm2[3]。并且随着经济的发展,我国的粮食安全还面临着几个重大的挑战,例如气候变化、土壤流失、病虫害问题等自然因素和人口数量的急剧增加以及多种因素共同影响下的可耕地面积的不断缩减等人为因素,这些都是造成小麦产量供不应求的原因。因此,为了满足我国日益增长的小麦需求,确保粮食安全,选育优质、高产、稳产的小麦品种成为小麦育种的主要目标之一[4]。提高小麦产量的策略主要有提高单位面积穗数、增加穗粒数、提高千粒质量等,三雌蕊小麦(TP)就是通过提高穗粒数,达到提高产量的目的。三雌蕊小麦(TP)是由Peng 等[5-6]对“三粒小麦”经多年的改良培育所得,TP是由一对显性核基因控制,与细胞质遗传无关。Yang等[7]已经利用基因分型技术(GBS)构建了小麦的高密度遗传图谱,并将控制小麦三雌蕊性状的Pis1基因定位在SNP标记M70与M71之间,与M70的距离为3.0 cM,与M71的距离为1.1 cM。
随着DNA分子标记技术的不断成熟与发展,利用分子标记进行新品种的选育已代替传统育种方式成为一种比较前沿的方法。分子标记的种类很多,如简单重复序列(SSR)、酶切扩增多态性(CAPS)、限制性片段长度多态性(RFLP)、随机扩增多态性DNA(RAPD)、单核苷酸多态性(SNP)和插入缺失标记(InDel)等[8-11]。其中SNP/InDel作为一种新型的分子标记技术,相比于其他类型的分子标记,有其本身特有的优势。InDel多态性是一种二等位基因遗传标记[12],可通过其表现形式分为以下5类:单碱基对的插入/缺失;单一碱基的插入/缺失;重复单元为2~15碱基的多碱基对插入/缺失;转座子插入/缺失;任意DNA序列的插入/缺失多态性。单核苷酸多态性(SNP)是指基因中单个碱基对发生替换、缺失或增添而引起的碱基序列改变,最终会导致DNA序列多态性。SNP标记具有位点多、分布广、易实现分析的多样化、稳定性较高、代表性强等特点。SNP标记在小麦育种中主要用于构建高密度的遗传连锁图、分子标记辅助育种、全基因组关联分析和物种进化等[13]。
本研究以三雌蕊近等基因系CM28TP与其轮回亲本川麦28(CM28)为试验材料,利用转录组测序(RNA-Seq)技术,对小麦幼穗的3个阶段进行测序,获得小麦SNP/InDel标记并进行生物信息学分析,将3个阶段共有的SNP/InDel位点所在的基因进行GO注释分析,获得有重要价值的SNP/InDel分子标记,为分子标记辅助选择育种和图位克隆控制小麦三雌蕊性状的Pis1基因奠定基础。
1 材料和方法
1.1 试验材料
本研究选用小麦三雌蕊近等基因系CM28TP与其轮回亲本川麦28(CM28)为试验材料,CM28是由四川省农业科学院作物研究所杨武云研究员提供。当小麦进入孕穗期后,采集长度为0.2~0.5 cm,0.5~0.7 cm,0.7~1.0 cm 3个阶段的小穗,浸入样品保护剂Sample Protector(TaKaRa,中国大连),液氮速冻后,于-80 ℃冰箱中保存备用。
1.2 转录组数据来源
由北京诺禾致源科技股份有限公司利用Illumina HiSeq PE150测序平台对CM28TP和CM28的幼穗(幼穗长度为0.2~0.5 cm,0.5~0.7 cm,0.7~1.0 cm 3个阶段)进行RNA-Seq测序,每个样本测3个生物学重复。测序获得的原始数据(Raw data)中包含少量带有测序接头或测序质量较低的数据(reads)。为了保证数据分析的质量及可靠性,需要对原始数据进行过滤。主要包括去除带接头的reads、去除含N(N表示无法确定碱基信息)的reads、去除低质量 reads(Qphred ≤20 的碱基数占整个 read长度的 50%以上的 reads),从而获得高质量序列(Clean reads)。同时,对Clean data 进行 Q20、Q30 和 GC 含量计算。后续所有分析均是基于Clean data进行。以中国春基因组(IWGSC1.0,ftp://ftp.ensemblgenomes.org/pub/plants/release-48/fasta/triticum_aestivum/dna/)为参考基因组,利用HISAT2 v2.0.5对Clean reads进行拼接。
1.3 SNP/InDel位点检测及GO分类分析
使用 GATK(3.7)软件对样本转录组数据进行SNP和InDel位点分析,为了获得准确的SNP和InDel位点信息,分别比较了CM28和CM28TP在幼穗不同发育阶段(幼穗长度为0.2~0.5 cm,0.5~0.7 cm,0.7~1.0 cm)的SNP和InDel位点,选择在CM28和CM28TP 3个幼穗发育阶段共有的SNP和InDel位点用于分析。利用SnpEff(4.3q)软件对变异位点进行注释。利用agriGO[14](http://systemsbiology.cau.edu.cn/agriGOv2/index.php)对CM28和CM28TP 3个幼穗发育阶段共有的SNP和InDel位点所在的基因进行GO分类分析。
1.4 SNP/InDel位点验证
控制小麦三雌蕊性状的Pis1基因位于2D染色体上,其物理位置为2D:580 226 867~601 216 125 bp[7,15]。因此,选择Pis1基因所在物理区间附近的4个SNP位点进行验证,利用Primer Premier 5.0 设计引物,SNP位点要尽量位于扩增片段的中间,引物信息如表1所示。PCR反应体系为25.0 μL,包括:2×TSINGKE Master Mix 12.5 μL;10 μmol/L的正向引物(F)和反向引物(R)各0.5 μL;模板DNA 2.0 μL;ddH2O 9.5 μL。PCR程序为:94 ℃预变性3 min;94 ℃变性30 s,52~60 ℃退火60 s(具体温度根据引物而定),72 ℃延伸1 min,共35个循环;最后72 ℃继续延伸5 min。PCR扩增结束后利用1%的琼脂糖凝胶进行检测,对PCR扩增产物进行回收纯化之后,连接至PMD-19T载体上,将连接产物转化至DH5α感受态细胞中,涂布于含有100 μg/mL氨苄青霉素的LB固体培养基上,37 ℃培养12~14 h,每个样品挑选10个PCR检测为阳性的克隆送至生工生物工程(上海)股份有限公司进行菌液测序,利用Chromas 2.0和DNAMAN 7.0对测序结果进行分析。

表1 位于Pis1定位区间附近的SNP标记Tab.1 SNPs near the Pis1 location
2 结果与分析
2.1 RNA-Seq数据质量评估
为了在转录水平上挖掘三雌蕊小麦和单雌蕊小麦之间的SNP位点,使用Illunina HiSeq PE150平台对来自CM28TP和CM28的0.2~0.5 cm(阶段1),0.5~0.7 cm(阶段2),0.7~1.0 cm(阶段3)的幼穗进行了转录组测序分析。对每个样品重复3次,测序读取长度为150 bp。在从原始reads中剔除接头序列、低质量reads和含有寡聚N的reads之后,平均Clean reads 为45 881 590.33,平均数据量为6.88 Gb,3个阶段的平均GC含量差值较小;Q20均大于97%;Q30均大于94%(表2)。表明测序结果良好,可以进行后续的SNP/InDel位点的数据挖掘与分析工作。

表2 RNA-Seq数据质量评估Tab.2 RNA-Seq data quality assessment
2.2 三雌蕊小麦转录组SNP/InDel类型分析
对CM28和CM28TP幼穗3个阶段的转录组数据进行比较分析,幼穗长度为0.2~0.5 cm的阶段获得SNP/InDel位点数共13 086个,幼穗长度为0.5~0.7 cm的阶段获得SNP/InDel位点数共11 001个,幼穗长度为0.7~1.0 cm的阶段获得SNP/InDel位点数共13 086个,3个阶段共有的SNP/InDel位点共5 310个,SNP/InDel发生频率为1/1 295 668,变异位点以SNP为主,共计5 024个;InDel位点286个,以2~10 bp的插入缺失为主。其中转换和颠换分别占63.33%,31.28%,两者的比值为2.02,286个变异位点超过2个核苷酸变异,约占5.39%(表3),转换类型中A/G型最多,颠换类型中C/G型最多。286个InDel类型中,有152个插入,134个缺失。
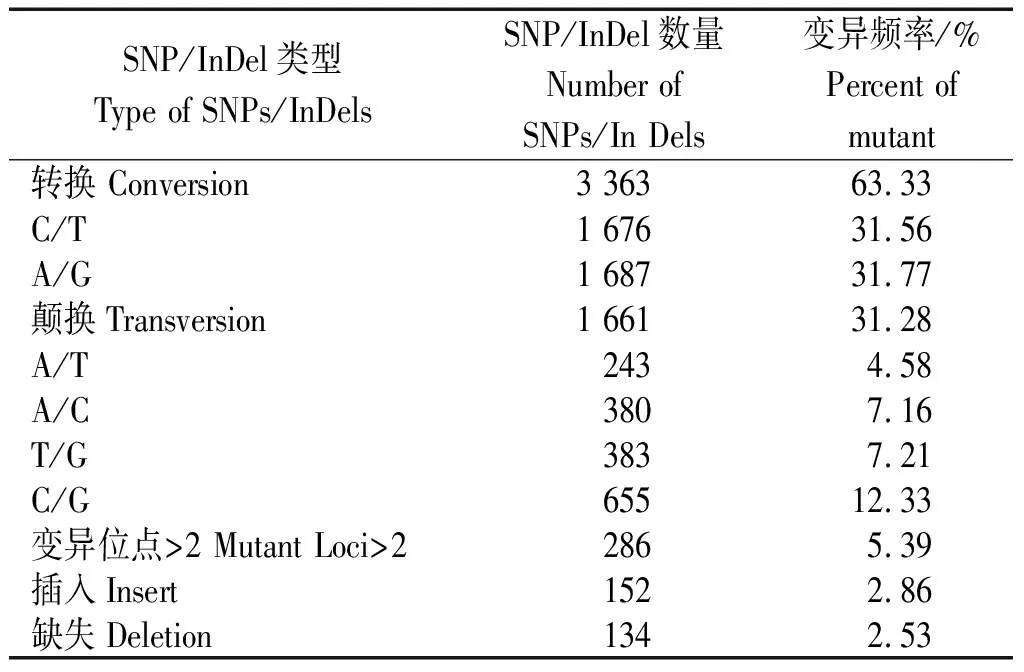
表3 SNP/InDel类型统计Tab.3 SNP/InDel type statistics
5 310个SNP/InDel中有4 543个是属于编码蛋白,占比85.55%;11个假基因,占比0.21%;18个核仁小RNA,占比0.34%;3个小核RNA,占比0.06%;其中还包括了735不确定编码类型的突变,占比13.84%(表4)。

表4 编码类型Tab.4 Encoding types
对筛选所得的5 310个SNP/InDel变异位点利用SnpEff(4.3q)软件预测其对编码蛋白进行功能性预测分析,将影响程度从高到低依次分为High、Moderate、Low和Modifier 4个等级。结果如图1所示。High代表严重影响蛋白质功能,共有36个(0.68%),Moderate代表中度影响蛋白质功能,共有1 279个(24.09%),Low表示对蛋白质功能的影响程度较低,共有1 849个(34.82%),Modifier表示与蛋白质的功能无关,共有2 146个(40.41%)。
对5 310个变异类型进行统计,结果如图2所示,其中1 824个属于同义突变,占比最高,达到了34.35%,1 221个错义突变,占比为22.99%,14个移码突变,占比0.26%,分布在3′端非编码区突变的位点747个(14.07%),分布在基因间隔区的位点735个(13.84%),分布在下游基因突变的位点252个(4.75%),分布在上游基因突变的位点182个(3.43%),另外还有一些占比较少的变异类型,如:43个内含子突变位点,30个保守框变异位点,26个破坏框变异位点,16个5′端过早起始密码子增益突变位点,6个终止增益变异位点,6个终止缺失变异位点,1个起始缺失变异位点,1个剪切区突变位点,19个SNPs位点被预测为鉴于2种变异类型之间。
2.3 SNP/InDel位点在染色体上的分布
5 310个SNP/InDel位点中有2 701个位于A基因组上,占比50.87%;1 912个位于B基因组上,占比36.01%;587个位于D基因组上,占比11.05%;110个SNP/InDel位点不能确定所在基因组,占比2.07%。其中分布在7A染色体上的SNP/InDel数是最多的,达到了774个;分布在4D染色体上的数量是最少的,仅有25个(图3)。
2.4 含有SNP/InDel位点的基因GO分类注释
GO分析有助于理解基因背后所代表的生物学意义。利用agriGO对CM28和CM28TP 3个幼穗发育阶段共有的SNP和InDel位点所在的基因进行GO分类分析。结果显示,5 310个SNP/InDel位点所在的基因主要分为3个大类和42个亚类(图4),占比
最高的是生物学进程,其次是细胞组分,最后是分子功能。生物学进程分为20个小类,细胞进程和代谢进程的基因数目最多;细胞组分分为12个小类,细胞部分和细胞的基因数目最多;分子功能分为10个小类,催化活性和结合的基因数目最多。
2.5 CM28与CM28TP中的SNP变异位点验证
从Pis1基因定位的物理区间附近选择4个SNP位点进行验证。这4个SNP位点分别命名为:T589505891C、G601219738A、A602512273G和T601058111C。其中T589505891C是2D染色体589505891位点由T突变为C,G601219738A是2D染色体601219738位点由G突变为A,A602512273G是2D染色体602512273位点由A突变为G,T601058111C是2D染色体601058111位点由T突变为C。经过PCR扩增和测序,发现这4个SNP位点在CM28和CM28TP中是真实存在的(图5)。因此,这4个位点可用于后续Pis1基因的定位。
3 讨论
小麦是世界上最常见的食用谷物,随着世界人口的快速增长,小麦生产将在粮食安全和全球经济中发挥举足轻重的作用。近年来,各种分子标记被广泛应用于小麦研究,如:侯起岭等[16]利用STS标记、SSR标记和KASP标记等方法鉴定小麦光温敏雄性不育系穗发芽抗性;朱靖环等[17]利用SSR标记方法筛选小麦抗赤霉病优异种质;魏广辉等[18]利用SNP开发出的KASP标记筛选高硒小麦。过去常用的其他分子遗传标记(SSR、CAPS、RFLP、RAPD)存在着开发成本高、重复性不好、数量有限等缺点,而新发展起来的SNP/InDel标记相对来说具有很多优势,比如在基因组中分布广、数量多、开发成本低、重复性好等,另外SNP/InDel 标记可用于分子标记辅助选择育种(MAS)、基因型分型、种间亲缘关系分析等[19]。SNP一般是指单个核苷酸的转换、颠换、插入和缺失,其中转换和颠换较为普遍,插入和缺失的情况较少;而InDel标记本质上是属于长度多态性标记,是指基因组中脱氧核苷酸的插入或缺失。本研究通过对小麦三雌蕊近等基因系CM28TP及轮回亲本CM28幼穗的3个阶段进行转录组测序,共挖掘出SNP/InDel位点5 310个,SNP/InDel发生频率为1/1 295 668,远低于其他物种。导致SNP/InDel发生频率低主要有以下两方面的原因:首先,本研究分析的是CM28TP和CM28小穗发育的3个阶段共有的SNP/InDel位点,有效地去除了假阳性位点,后续在Pis1基因的定位区间附近选择了4个位点进行验证,也证实这4个位点在CM28TP和CM28中真实存在。这也进一步佐证了挖掘出的5 310个SNP/InDel位点的准确性。其次,CM28TP和CM28为近等基因系,其遗传差异本身较小[20]。
在5 310个SNP/InDel位点中碱基变异类型主要为C/T、A/G,以转换为主,转换和颠换的比值为2.02,远大于理论值0.5。生物中SNP位点碱基变异的转换和颠换的比值往往会大于理论值0.5,这种现象称为“转换偏差”[21]。王妍等[22]对向日葵锈菌的转录组数据分析结果中,转换发生的频率是65.40%,颠换发生的频率为34.60%;陈姝欣等[23]对于人参果转录组测序结果中,转换类型发生的频率为61.10%,颠换类型发生的频率为38.27%;张雨等[24]对山茛菪转录组数据进行分析,结果显示,转换类型占64.26%,颠换类型占35.74%。这一现象说明,SNPs/InDels位点的碱基突变可能会受环境等其他因素的影响,并不是随机发生的。在自然选择过程中,转换突变在蛋白编码序列中会产生同义突变,因此,通常情况下SNP的转换类型出现频率都高于颠换类型。从SNP/InDel位点在染色体上的分布来看,A基因组上的位点最多,达到50.87%,其次是B基因组,为36.01%,D基因组上的位点最少,仅11.05%。这可能与D染色体组在进化上比较保守有关[25-26]。
经过GO分类注释,5 310个SNP/InDel位点所在的基因主要分为3个大类和42个亚类,其中细胞进程、代谢进程、细胞成分、细胞、催化活性和结合这几个亚类所包含的基因最多。这些亚类中所包含的基因可能在植物生长发育中发挥了重要的功能,但这些基因在小麦三雌蕊性状发育中的作用还有待进一步研究。本研究从Pis1基因定位区间附近挖掘出4个SNP位点,经过PCR扩增和测序验证,这4个位点与RNA-Seq结果一致,下一步将在CM28×CM28TP的重组自交系(RILs)群体中验证这4个SNP位点,并计算其与Pis1基因之间的遗传距离,进一步精细定位Pis1基因。
近几年来,SNP标记在小麦的遗传育种工作中受到了广泛的关注,SNP标记与第一、二代标记相比,具有位点多、分布广、稳定性高和代表性强等特点,所以具有更大的使用价值和发展空间。在小麦的遗传育种工作中,将SNP用于高密度遗传连锁图谱的构建,更有利于基因的精细定位,从而为小麦的基因组分析、表型变异研究以及物种资源鉴定提供新途径;其次,将SNP标记技术作为选择育种的辅助手段,将极大地提高育种的可预测性和选择性,从而使新品种选育的判断更具有客观性;除此之外,通过分析小麦基因中存在的SNP,可以了解其物种进化过程中在基因组水平上的DNA多态性,从而分析出不同小麦群体的结构分类、遗传多样性或者群体分化等特点。简言之,SNP标记技术将有利于小麦遗传育种工作的进行,是小麦农艺性状研究的一大助力[13]。
