高压液相色谱-荧光检测法检测蜂王浆中喹诺酮类药物残留的方法
2021-10-23王圣义
王圣义,胡 娟
(1.武汉市华测检测技术有限公司,湖北 武汉 430070;2.湖北省扬子江蜂业有限公司,湖北 武汉 430070)
诺氟沙星、环丙沙星和恩诺沙星均属氟喹诺酮类,为喹诺酮类第三代药物,属于广谱抗菌药[1-3]。在养蜂生产过程中,蜂农为了预防和治疗蜂病,有时要用到药物,而药物的来源很混杂,有人用药和兽用药,还有的蜂农使用了一种含有喹诺酮类药物成分却没有进行标签标识的药物,这样就可能导致生产的蜂王浆中有喹诺酮类药物残留。
国内外检测喹诺酮药物残留的方法有很多,主要有微生物法[4-6]、酶联免疫法[7-9]、高压液相色谱法[10-13]、液相串联质谱法[14-16]、毛细管电泳法[17-18]等。目前在蜂产品行业,特别是蜂王浆生产企业,大多以酶联免疫方法作为蜂王浆兽药残留监控的主要手段。酶联免疫方法具备快速简便、灵敏度高、特异性强、对设备要求不高以及检测成本低等特点,但酶联免疫法不能将每种喹诺酮类药物单独检验而准确定量。高压液相色谱仪在一定程度上可以实现对不同喹诺酮药物的分离而进行单个定量,这对于蜂王浆生产企业而言有重要的意义。因此,对蜂王浆中喹诺酮类药物残留量的检测方法进行研究,对于丰富兽药残留的检测方法以及增强企业自检自控能力非常有意义。
1 材料与方法
1.1 仪器和试剂
DIONEX U3000 高压液相色谱仪配RF2000 荧光检测器(美国戴安公司),SHZ-82A 水浴恒温振荡器(金坛区金南仪器厂),PHS-3G 高精度pH 计(上海利达仪器厂),TGL-16C 台式离心机(上海安亭科学仪器厂),固相萃取装置(安捷伦科技有限公司)。
乙腈(ACN)、甲醇、甲酸均为色谱纯,柠檬酸、乙酸铵、乙酸乙酯、氢氧化钠、盐酸、磷酸二氢钾、三乙胺均为分析纯,三氟乙酸(TFA) 为化学纯;柠檬酸-乙酸铵混合盐溶液:分别精确称取10.507 0 g 柠檬酸、0.770 8 g 乙酸铵,用重蒸馏水溶解并稀释定容至1 000 mL,用三乙胺调节pH 为3.0;诺氟沙星、环丙沙星和恩诺沙星标准品(德国Dr.Ehrensorfer 公司)

1.2 实验方法
1.2.1 样品处理
准确称取试样2 g,精确至0.01 g,置于100 mL塑料瓶中,加入0.2 mol/L 的三氟乙酸溶液10 mL,于漩涡混合器上将样品分散,然后加入10 mL 乙腈,于康氏振荡器上振荡20 min,然后转移至50 mL 的塑料离心管中,以4 000 rpm 离心8 min,然后将上清液转移至100 mL 的茄型瓶中,再向沉淀中加入10 mL的0.2 mol/L 的三氟乙酸溶液,于漩涡混合器上将沉淀分散,然后转移至100 mL塑料瓶中,再加入10 mL 乙腈,于康氏振荡器上振荡10 min,再以4 000 rpm /min离心8 min,合并两次的上清液,将上清液于50℃旋转蒸发除去乙腈,然后将经过浓缩的样液于15 000 rpm/min 离心5 min。上清液备用。
将SCX 固相萃取小柱安装在固相萃取装置上,分别先后用1 柱管甲醇和1 柱管水活化柱子。然后将样品处理得到的上清液转移至SCX 柱上,通过调节真空度让样液以4 秒1 滴的速度通过柱子。待样液全部通过柱子后(注意保持柱体湿润),先用2 柱管水以2~3 秒1滴的速度淋洗柱子,再用1 柱管甲醇和1 柱管水以同样速度淋洗柱子。最后用4 mL 0.1mol/L氢氧化钠溶液洗脱柱子,收集洗脱液。向洗脱液中添加200 μL 15%的 柠檬酸,混匀,用0.45 μm 的滤膜过滤,待测。
1.2.2 标准溶液的配制
用十万分之一电子天平称取诺氟沙星、环丙沙星和恩诺沙星标准品约0.003 00~0.005 00 g 到10 mL容量瓶中,加入0.1 mol/L 氢氧化钠溶液2 mL和甲醇5 mL,超声使之溶解,静置,用甲醇定容。然后各移取适量溶液至另一容量瓶中,制成浓度为100 μg/mL 贮备液,存放于4℃冰箱中。
移取标准贮备液0.5 mL,用甲醇稀释并定容于50 mL容量瓶中,制成浓度为1 μg/mL的标准工作液。使用时用流动相稀释标准工作液至所需浓度。
(3)规划水平年(2020年、2030年)灌溉保证率50%相比75%,水资源承载能力评分较高,说明可以采用降低保证率灌溉更多耕地的方法以达到总产值的增加,提高山塘水资源的承载能力。
1.2.3 仪器条件


2 结果与分析
2.1 色谱条件的选择
2.1.1 色谱柱及检测器、检测波长的选择
喹诺酮类物质属于酸碱两性物质,其结构中的叔胺基和羟基官能团能在水中发生解离,色谱柱固定相表面残存的硅醇基和金属离子可以通过氢键或离子交换作用强烈吸附喹诺酮类化合物而出现峰形变宽、拖尾以及保留时间不稳定或过长,甚至被保留在色谱柱上,导致峰形异常和分离度下降[19]。因此需选用高纯硅胶为基质并经过封端处理的C18 等反相色谱柱作为分离柱。实验在所使用的几款色谱中,发现美国戴安公司生产的ACCLAIMTM120 C18 柱(5 μm,120 ,4.6 mm(id) ×250 mm) 能将3 种氟喹诺酮类药物很好分离,并且峰形对称而且尖锐,该色谱柱耐高压且能长期使用,完全符合试验要求。
喹诺酮类药物本身具有较强的荧光吸收,无需进行衍生化就能直接进行分析,而且检测灵敏度高,因此实验选用荧光检测器。通过荧光波长扫描,以及参考他人研究方法[20],选择RF2000 型荧光检测器,激发波长280 nm,发生波长450 nm,可调节灵敏度和电信号增益,本次实验采用的模式为“gain:4,sensitivity:high”。
2.1.2 流动相的选择
喹诺酮类药物具有可质子化的氮原子和离解的羟基,采用反相色谱柱测定此类药物时,会出现峰的拖尾现象,必须在流动相中加入离子对试剂。离子对试剂包括磷酸盐缓冲盐溶液(三乙胺调节pH 值) -乙腈、四丁基溴化铵溶液- 乙腈、乙腈-(柠檬酸、醋酸铵混合溶液) 等。
比较了乙腈-(柠檬酸、醋酸铵混合溶液) 以及0.01 mol/L 四丁基溴化铵溶液(75%磷酸调节pH 为3) - 乙腈。发现采用乙腈-(柠檬酸、醋酸铵混合溶液) 作为流动相时,不能将诺氟沙星和氧氟沙星分开,而采用0.01 mol/L 四丁基溴化铵溶液(75%磷酸调节pH 为3) - 乙腈可以将诺氟沙星和氧氟沙星分开,但是氧氟沙星的检出限偏低,考虑到研究的目的,最终选择了乙腈-(柠檬酸、醋酸铵混合溶液) 作为流动相。结合使用戴安 公 司 的ACCLAIMTM120 C18 柱(5 μm,120 ,4.6 mm(id) ×250 mm),得到了较好的分离效果以及峰形,见图4。
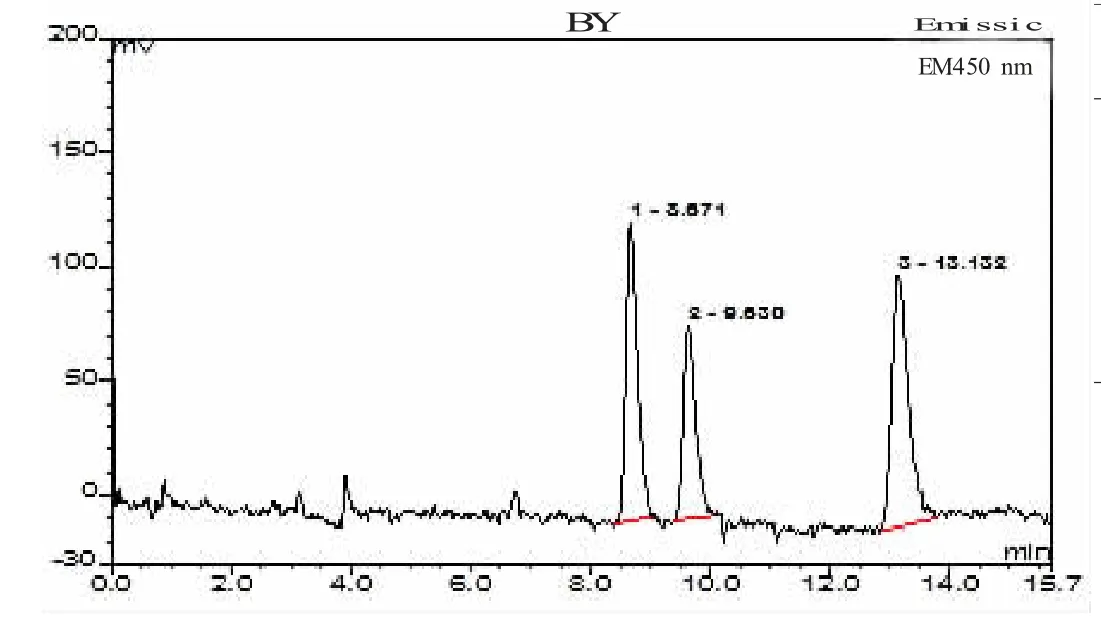
图4 标准品色谱图Fig.4 HPLC Chromat ogram of Mixed St andard QNs
2.2 样品前处理条件的优化
2.2.1 样品提取条件的优化
由于蜂王浆中成分复杂,含有大量的蛋白质、脂肪、糖类等物质,而不能通过采用简单的稀释处理而直接进样分析。周萍等[21]研究了通过液液提取净化的方法,由于受净化方法的影响,通过净化后的试样氟喹诺酮类药物残留有一定的损失,样品回收率范围在46.5%~55.9%,通过采用基质标样以及添加内标的方法对结果进行了校正。Lambardo-Aguei M 等[22]在采用液相串联质谱法检测蜂王浆的QNs 时为降低基质的干扰,通过减少称样量的办法以及应用到了固相萃取净化方法。本研究没有采用基质标样,而是直接采用标准工作液进行分析。在样品前处理方面,通过采用合适的提取试剂以及利用固相萃取技术和合适的洗脱方式对样品进行了净化。
实验比较了乙酸-乙腈体系、三氟乙酸-乙腈体系、偏磷酸-乙腈体系分别作为提取剂的效果。结果发现,使用三个体系都可以将三种氟喹诺酮类药物从蜂王浆中提取出来。而三氟乙酸-乙腈体系优于另外两种提取体系。
在对三氟乙酸-乙腈体系进行研究时,比较了不同pH 值分别为1.07、3、4.2 的三氟乙酸水溶液与乙腈组成的提取效果与基质干扰物的去除效果,结果表明pH 为3 的提取液基质干扰物含量少,最终选择pH 3.0、0.2 mol/L三氟乙酸和乙腈组成的提取液体系。三者的提取效果(5%氨水甲醇洗脱物) 见图1。图中显示在RT 为8.5 、 9.5、13.0 min 时,均不同程度存在干扰。在RT=9.5 时,1#有强干扰物存在。

图1 不同pH值三氟乙酸- 乙腈对空白样品基质的提取效果比较Fig.1The comparison of ext ract ing effect s on t he mat rix of blank samples by TFA- ACN at different pH value
2.2.2 提取液净化方法的选择
通过比较C18 固相萃取小柱、AX 固相萃取小柱以及SCX 小柱对样品的净化效果发现:如果按小柱所提供的说明书操作,一般得到的色谱图中含有大量的基质干扰,见图2。后来通过对SCX 柱的净化方法进行探索,发现采用5%的氨水甲醇对SCX 小柱进行洗脱,只能洗脱目标物的30%~50%,而且洗脱液在经氮气吹干后的复溶液经过仪器检测发现含有大量干扰物质,见图2。为了能将目标物从SCX 小柱上完全洗脱下来,实验研究了不同洗脱液的洗脱效果,结果发现使用4mL 0.1mol/L 的氢氧化钠溶液可以洗脱90%的被吸附的氟喹诺酮类药物。而采用200 μL 15%的柠檬酸调节洗脱液pH 值后,经过仪器进行分析发现在药物色谱保留时间,空白样品几乎没有基质干扰,见图3。
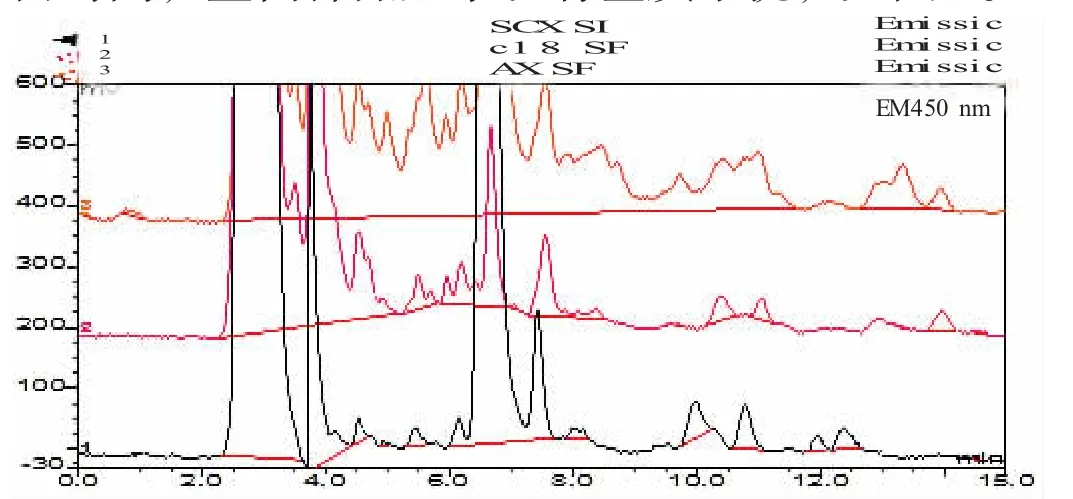
图2 三种固相萃取小柱对空白蜂王浆的净化效果比较Fig.2 Comparison of clean- up effect s on blank royal j elly of t hree kinds of SPE columns
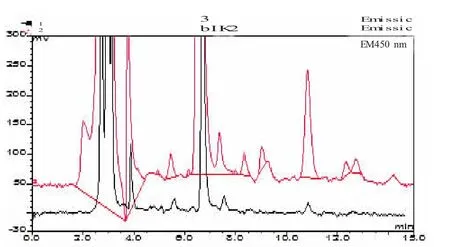
图3 两种针对SCX吸附空白蜂王浆基质洗脱效果的比较Fig.3 comparison of t wo elut ingeffect s on mat rix of blank royal j ellyabsorbed by SCX SPE column
2.3 标准曲线
按照标准曲线的绘制方法进行对标准样品的测试,记录色谱峰高。诺氟沙星、环丙沙星和恩诺沙星在2~50 μg/kg 范围内线性良好,当浓度达到100 μg/kg 时,在选定色谱条件下浓度信号值超出了仪器的测定范围。以诺氟沙星、环丙沙星和恩诺沙星的峰高对诺氟沙星、环丙沙星和恩诺沙星浓度作回归分析,线性回归方程和相关系数见表1。空白蜂王浆样品以及空白添加回收色谱见图5、图6。
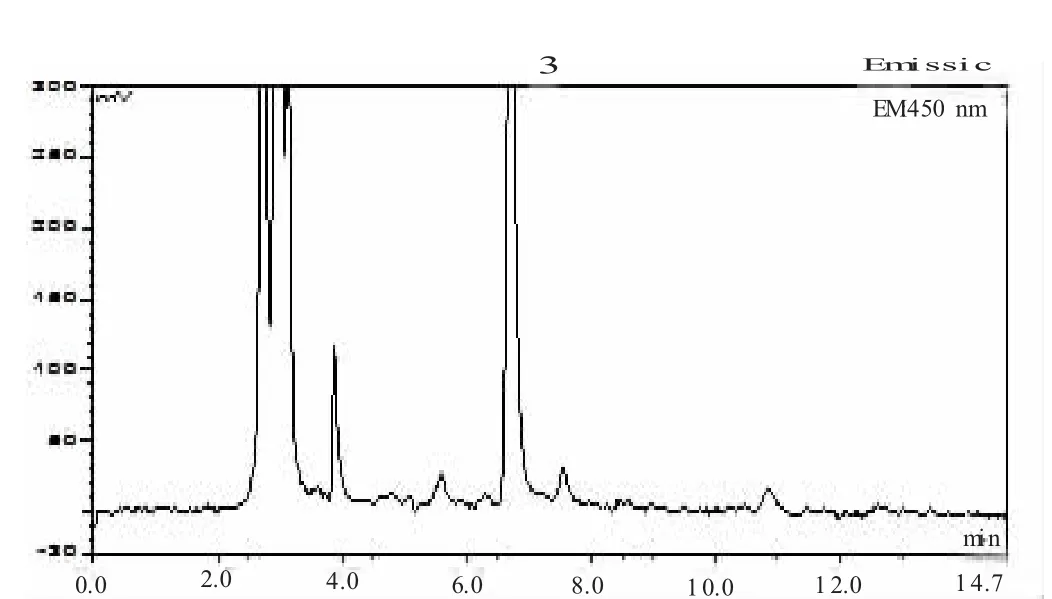
图6 空白蜂王浆色谱图Fig.6 HPLC Chromat ogram of blank royal j elly
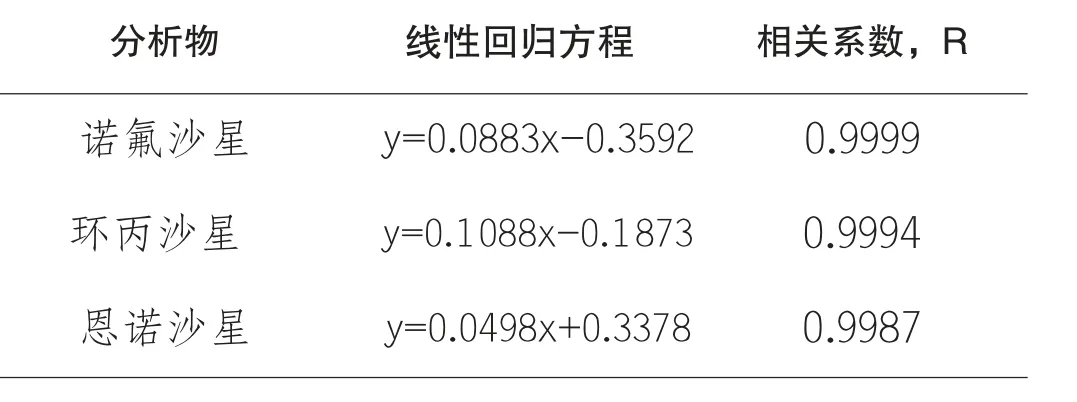
表1 3 种氟喹诺酮类药物的线性回归方程及相关系数Table 1 Linear regressive equat ions and correlat ion coefficient s for t hree FQNs
2.4 方法的回收率和精密度测试
检出限是指能够被检测到的分析物的最小含量,它与所使用的方法和设备有关。本实验研究所使用的RF2 000 型荧光检测器可调节灵敏度和电信号增益。本次研究采用的模式为“gain:4,sensitivity:high”,在这种模式下,样品基质的噪音也会被相应放大,但是却得到了比较好的目标分析物信号。回收率实验按照1 倍、2 倍和10 倍定量限三个浓度水平进行添加标准工作液。测得本实验方法的检测限为1μg/kg(S/N=3),定量限为3μg/kg(S/N≈10)。实验过程中称取空白蜂王浆2.0 g,分别添加3 种氟喹诺酮类药物含量为6.0 ng、12.0 ng、60 ng 标准到蜂王浆中,漩涡混合1min,置于暗处放置30 min,然后按本方法进行样品处理和检测,精密度测试结果为三种氟喹诺酮药物残留含量在各个水平的RSD 值均低于20%。结果见表2,回收率和精密度均符合GB/T 27404-2008《实验室质量控制规范食品理化检测》标准对检测方法确认的技术要求的规定。
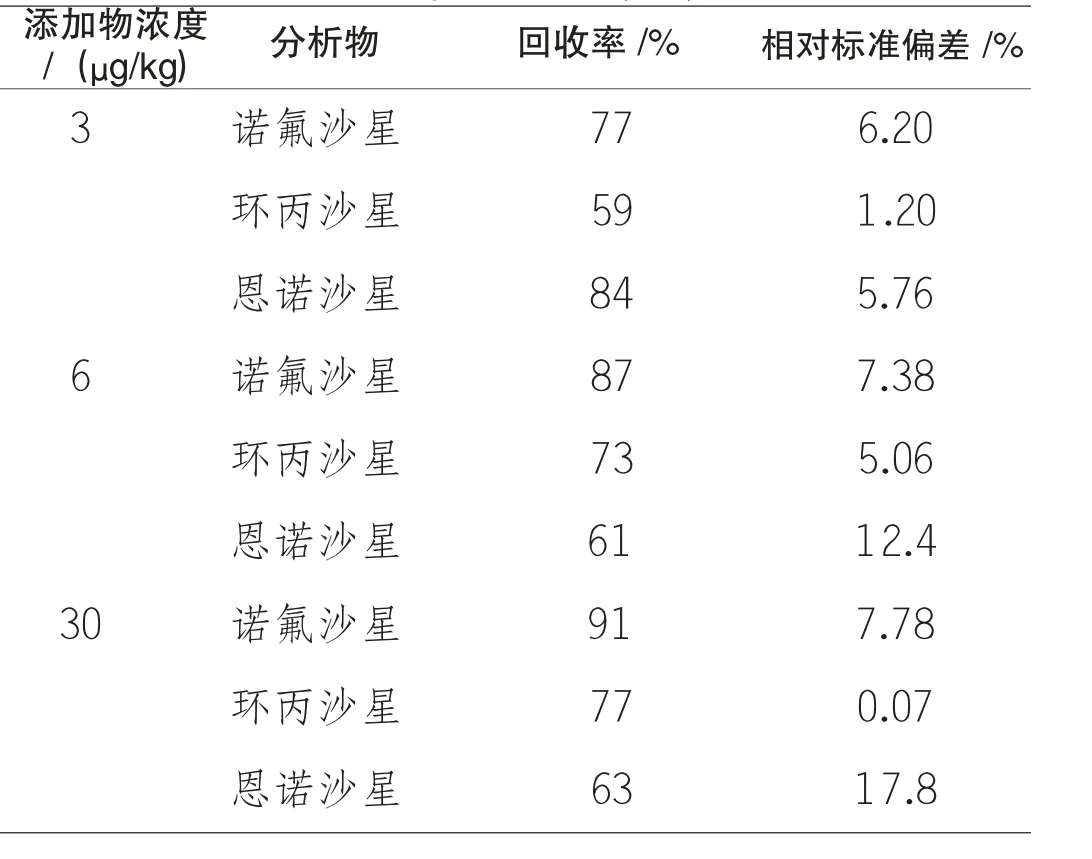
表2 3 种氟喹诺酮类药物残留的加标回收率与相对标准偏差(n=5)Table 2 Spiked recoveries and relat ive st andard deviat ions of 3 QNs residues(n=5)
3 结论
本试验研究了不同提取液、不同SPE 柱、不同洗脱液对测试结果的影响,最终建立了以三氟乙酸- 乙腈提取蜂王浆中的3 种氟喹诺酮药物残留,提取液经过浓缩后,经过SCX 固相萃取小柱进行样品净化,最后用4mL 0.1 mol/L 氢氧化钠溶液洗脱,再用15%柠檬酸调节pH 值,然后经过高压液相色谱- 荧光检测器检测的方法。在该方法中,采用氢氧化钠洗脱和用柠檬酸调节pH 值的试样液可以最大限度地降低蜂王浆基质的影响。方法具有检测限低、准确度高等特点,可以作为蜂王浆生产企业和检测机构进行蜂王浆3 种氟喹诺酮的残留检测方法。
