Good综合征诊治一例
2021-10-20陈珊珊郭子文叶永斌苏冰源黄贵年
陈珊珊 郭子文 叶永斌 苏冰源 黄贵年


【摘要】Good综合征(伴胸腺瘤的免疫缺陷症)是一种成人免疫缺陷病,特点是合并胸腺瘤及低丙种球蛋白血症。该病有细胞和体液免疫双重缺陷,患者常表现为反复感染。该文报道1例62岁AB型胸腺瘤女性患者,反复细菌、真菌和病毒感染,血清Ig低下,B淋巴细胞缺如、CD4+ T淋巴细胞下降、CD4+/CD8+ T淋巴细胞比值下降,诊断为Good综合征。该患者同时有粒细胞缺乏、慢性病性贫血,进一步骨髓宏基因组测序发现KIT与ARID1A基因突变,予补充Ig及抗感染治疗,患者症狀好转出院。随访患者仍有Ig下降,予定时复查血常规、Ig,并适时补充Ig。Good综合征相对罕见,临床表现复杂,容易延误诊断,临床医师需引起重视。
【关键词】Good综合征;伴胸腺瘤的免疫缺陷症;免疫缺陷;胸腺瘤;粒细胞缺乏
Diagnosis and treatment of Goods syndrome: a case report Chen Shanshan, Guo Ziwen, Ye Yongbin, Su Bingyuan, Huang Guinian. Division of Hematology, Affiliated Zhongshan Hospital, Sun Yat-sen University,
Zhongshan 528403, China
Corresponding author, Huang Guinian, E-mail: hgn1997@163.com
【Abstract】Goods syndrome (immunodeficiency with thymoma) is an adult immunodeficiency disease characterized by thymoma complicated with hypogammaglobulinemia. Due to dual deficiency of cellular and humoral immune functions, patients constantly present with recurrent infection. This article reported one 62-year-old female patient with type AB thymoma presenting with recurrent bacterial, fungal and viral infection, low serum immunoglobulin, B lymphocyte deficiency, decreased CD4+ T cells and CD4+/CD8+ T ratio, which were consistent with the diagnosis of Goods syndrome. Meanwhile, the patient was complicated with agranulocytosis and anemia of chronic disease. Furthermore, bone marrow genetic detection revealed mutations in KIT and ARID1A genes. The patient was discharged after immunoglobulin supplement and anti-infection therapy. During follow-up, serum immunoglobulin level was declined. Routine blood test was performed on a regular basis. Immunoglobulin supplement was given when necessary. Goods syndrome is relatively rare in clinical practice. It is likely to delay the diagnosis due to complex clinical manifestations, which is worthy of widespread attention from clinicians.
【Key words】Goods syndrome; Immunodeficiency with thymoma; Immunodeficiency; Thymoma;
Agranulocytosis
Good综合征(伴胸腺瘤的免疫缺陷症)是罕见的成年发病的原发性免疫缺陷病,以胸腺瘤、低丙种球蛋白血症、CD4+/CD8+ T淋巴细胞比值下降(倒置)、低外周血B细胞和伴有CD4+ T淋巴细胞免疫缺陷为特点[1]。0.4% ~ 6.0%的胸腺瘤患者合并Good综合征,该病46.9%的病例出现在欧洲,其次为美国,国内尚无患病率报道[2-3]。Good综合征发病率低、临床表现复杂,容易误诊、漏诊[4]。为加深临床医师对该病的认识,现将笔者近期收治的一例Good综合征诊治过程报告如下。
病例资料
一、病史与体格检查
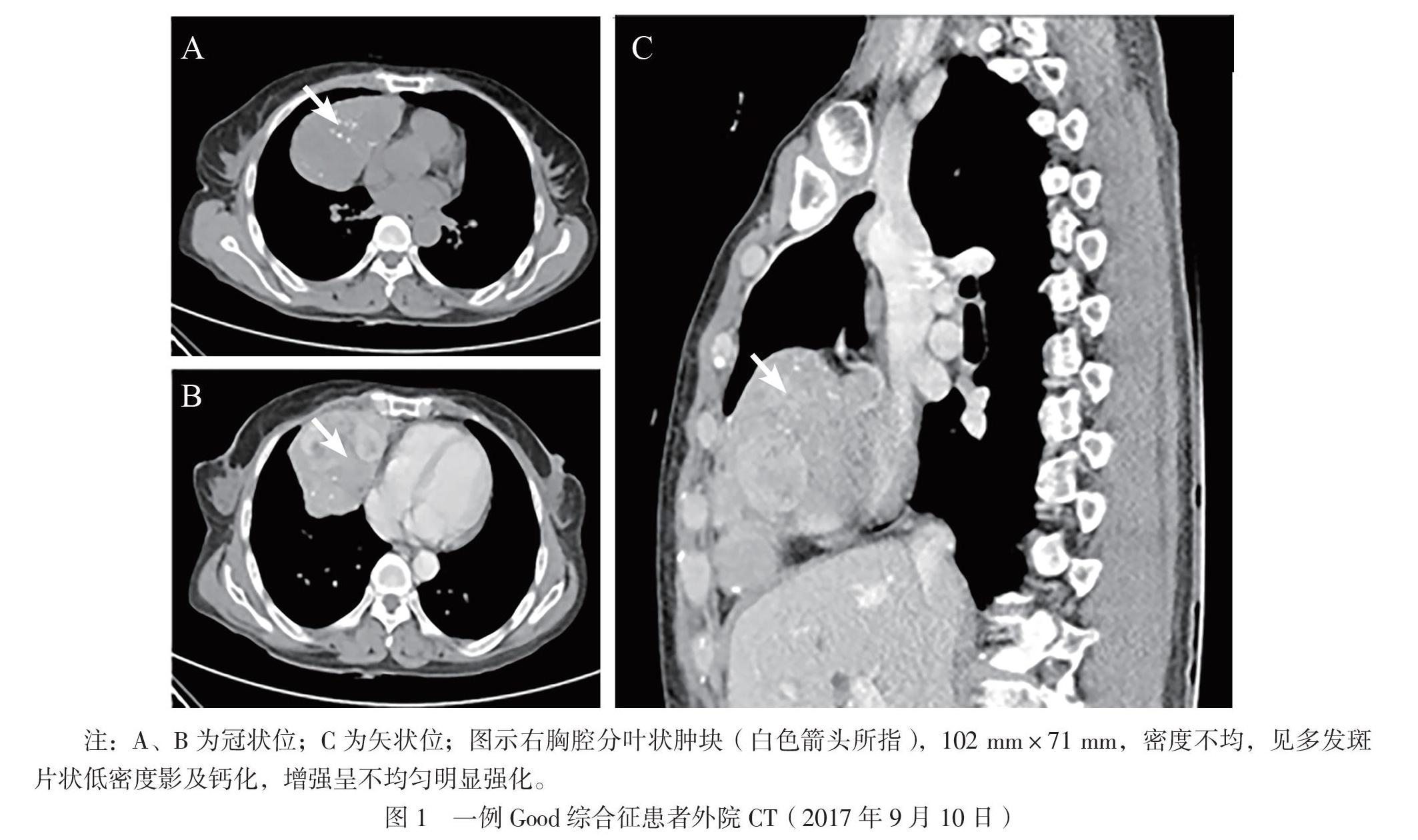
患者女,62岁。因胸部肿物15年,反复咳嗽、发热、乏力6年于2020年8月6日入院。患者于2005年8月CT检查发现胸部肿物,疑诊肺癌,放弃治疗。2014年8月患者出现咳嗽伴发热、乏力,查白细胞2×109/L,后出现手足“灰指(趾)甲”。2017年9月患者咳嗽加重伴呼吸困难,CT示右侧胸腔巨大分叶状肿块(图1);外院不排除周围型肺癌行手术治疗,病理示AB型胸腺瘤(图2)。术后患者呼吸困难缓解,其他症状反复,多次痰培养见革兰阳性链球菌、黄曲霉。2020年8月患者再次出现咳嗽、发热,体温最高39℃,CT示双肺支气管扩张并感染(图3A),在外院予抗感染治疗。后患者因全血细胞减少(血红蛋白81 g/L,白细胞 0.97×109/L,血小板96×109/L)就诊于我院。既往史:12年前患者左额面部带状疱疹,遗留神经痛。
体格检查:体温36.5 ℃,脉搏113次/分,血压88/65 mm Hg(1 mm Hg = 0.133 kPa),呼吸20次/分。贫血貌。双肺呼吸音粗,可闻及吸气相湿性啰音。肝肋下1 cm可触及,质稍硬,无压痛;脾肋下3 cm可触及,质地中等,无压痛。多个指(趾)甲可见变形、不规则、脱屑、发黄,双手掌、足趾可见白色脱屑。
二、实验室及辅助检查
患者入院血常规:血红蛋白82 g/L,红细胞2.93×1012/L,白细胞1.07×109/L,嗜中性粒细胞绝对值0.18×109/L,淋巴细胞绝对值0.47×109/L,红细胞平均体积81.9 fL,红细胞平均血红蛋白含量28 pg,红细胞平均血红蛋白浓度342 g/L 。网织红细胞计数升高(175.70×109/L)。患者反复感染的特点提示其免疫力低下,本中心结合其胸腺瘤病史并复习文献后考虑Good综合征的诊断可能。入院后进一步完善实验室检查,免疫学指标:血清Ig降低(IgA< 0.50 g/L,IgG< 3.00 g/L,IgM< 0.25 g/L)。骨髓流式细胞仪结果:未见明显成熟B淋巴细胞。外周血流式细胞仪结果:淋巴细胞计数明显下降,B淋巴细胞0 /μL (参考值范围74 ~ 534 /μL),CD3+ T淋巴细胞492 /μL (参考值范围711 ~ 2351 /μL),CD4+T淋巴细胞168/μL (参考值范围368 ~ 1632 /μL),CD8+ T细胞202/μL (参考值范围201 ~ 931 /μL),CD4+/CD8+ T淋巴细胞比值0.83 (参考值范围0.98 ~ 1.94)。
完善骨髓常规示:粒细胞成熟障碍伴胞浆颗粒增粗,外周血粒细胞比例低;骨髓活组织检查(活检)示:骨髓造血面积无减少,未见病态造血;这提示患者合并粒细胞缺乏。血常规提示正细胞性贫血,但患者脾大,网织红细胞计数升高,骨髓活检正常可排除再生障碍性贫血,完善自身抗体、中性粒细胞胞浆抗体、抗人球蛋白试验,酸溶血试验、蔗糖水试验、红细胞渗透脆性试验、尿含铁血黄素试验均(-),可排除自身免疫性贫血、溶血性贫血。进一步完善HBV标志物检查,结果示HBsAg (+)、HBeAg (+)、HBcAb (+)、抗-HBs (-)、抗-HBe (-);HBV DNA 5.61×108 copies/mL。肝功能白蛋白28.20 g/L,球蛋白13.90 g/L,白蛋白/球蛋白2.0,总胆红素41.40 μmol/L,直接胆红素14.70 μmol/L,间接胆红素26.70 μmol/L。
凝血功能活化部分凝血活酶时间 39.50 s,纤维蛋白原1.88 g/L;肝纤维化指标:Ⅳ型胶原 244.19 μg/L。CT:肝脾增大、脾大明显,胆总管轻度扩张(图3B)。患者胆红素升高、低蛋白血症、凝血异常、肝纤指标升高、肝脾肿大,结果提示患者乙型肝炎,不能排除肝炎后肝硬化、脾功能亢进导致红细胞及血小板继发性降低。
三、诊疗经过
结合病史及实验室检查,患者诊断Good综合征明确。同时1型辅助性T淋巴细胞/2型辅助性T淋巴细胞(Th1/Th2)亚群检测示IL-4 5.21 ng/L (参考值范围0 ~ 4 ng/L),IL-6 107.52 (参考值范围0 ~ 5.30 ng/L),IL-10 27.53 ng/L(参考值范围0 ~ 4.91 ng/L),不僅提示患者存在感染,同时表明Th2分泌的辅助B淋巴细胞分化的细胞因子代偿性增加,进一步证实了Good综合征中B淋巴细胞缺乏导致的体液免疫缺陷。患者甲(皮)屑真菌检查:见真菌菌丝、真菌孢子,同时患者肺部感染均提示了其为免疫力降低导致的机会性感染。
为进一步排查有无血液系统肿瘤及相关遗传性疾病,行造血与淋巴组织肿瘤基因突变检测(骨髓),结果提示KIT与ARID1A基因高频突变,ARID1A (NM006015):突变位置exon1,c.140G>A (p.R47H) ,突变率(测序深度)49% (813X);KIT (NM000222):突变位置exon2,c.185G>A (p.G62D) ,突变率(测序深度)49.1% (1661X)。骨髓增殖性肿瘤相关基因突变及融合基因检测(骨髓)未见异常(JAK2、MPL、CALR、CSF3R基因突变阴性,BCR-ABL1融合基因p210、p190、p230亚型均为阴性)。骨髓增生异常综合征基因突变检测(骨髓)阴性。血液肿瘤分子核型分析(骨髓)未检测到染色体拷贝数异常。遗传病全外显子组基因测序(全血)未见致病变异。
治疗上予Ig替代治疗(静脉注射人Ig,每日5 g连用3 d),治疗后复查IgG明显上升(IgG 6.45 g/L);予恩替卡韦抗HBV治疗;予特比萘芬治疗手足癣、甲真菌感染;入院后复查CT提示肺部感染较前明显减轻(图3C),未予抗肺部感染治疗。入院1周后患者咳嗽、发热等症状改善,2020年8月14日出院。出院后电话随诊,继续予恩替卡韦抗HBV治疗,但其未遵医嘱规律复诊进行血常规及Ig指标的检查。
患者出院7个月后(2021年3月26日)因发热、咳嗽、眼周疱疹返院完善检查。免疫学指标:IgG< 3.00 g/L。外周血流式细胞仪检测:CD4+/
CD8+ T淋巴细胞比值0.61,B淋巴细胞计数0 。痰培养见铜绿假单胞菌。CT提示支气管扩张并肺部感染(图3D)。考虑Good综合征Ig降低后免疫力低下再次导致机会性感染,予补充IgG治疗(静脉注射人Ig,每日5 g连用3 d),治疗后复查IgG上升至7.88 g/L。因先后予哌拉西林钠他唑巴坦钠、莫西沙星抗细菌及泛昔洛韦分散片、更昔洛韦眼用凝胶抗病毒、氟康唑片抗真菌治疗后患者仍反复发热(CRP 123.14 mg/L,血清淀粉样蛋白>200 mg/L,降钙素原0.14 μg/L),完善病原微生物高通量感染测序(全血)提示患者存在人类疱疹病毒Ⅰ型感染,疑似球形马拉色菌、近平滑念珠菌真菌感染。患者免疫缺陷,补充Ig后感染难以控制,考虑与其粒细胞缺乏有关,予重组人粒细胞刺激因子(每日250 μg连用 6 d)治疗后,白细胞、中性粒细胞可上升至正常,后患者发热缓解,复查CRP、血清淀粉样蛋白、降钙素原炎症指标下降。复查肝脏相关指标:HBV DNA病毒载量较前有所下降(3.67×108 copies/mL);肝功能白蛋白32.2 g/L,球蛋白17.6 g/L,白蛋白/球蛋白2.8,总胆红素19.30 μmol/L,直接胆红素7.30 μmol/L,间接胆红素12.00 μmol/L,提示肝功能好转。肝脏纤维化指标及凝血功能恢复正常,肝纤维化B超示肝纤维化硬度< 7.3 kPa,可排除肝硬化及继发性脾功能亢进。血常规:血红蛋白95 g/L,白细胞0.74×109/L,嗜中性粒细胞绝对值0.02×109/L,淋巴细胞绝对值0.41×109/L,红细胞平均体积78.4 fL,红细胞平均血红蛋白含量25 pg,红细胞平均血红蛋白浓度316 g/L,血小板193×109/L。网织红细胞计数正常(82.5×109/L)。血铁5项:血清铁8 μmol/L,转铁蛋白1.8 g/L,未饱和铁结合力26μmol/L,总铁结合力34 μmol/L,铁蛋白771 μg/L。骨髓常规:粒细胞成熟障碍,细胞外铁正常,内铁减少。患者贫血由正细胞性贫血转为小细胞低色素性贫血,提示其贫血为慢性病性贫血,患者血小板恢复正常,考虑之前降低与感染有关。本次出院嘱患者严格随访,每月复查血常规、Ig,并适时补充Ig,随访至撰稿日其IgG为5.98 g/L。
讨论
Good综合征患者除胸腺瘤外常见的临床表现是肺部感染,其次是腹泻、机会性感染、真红细胞再生障碍性贫血、口腔念珠菌病及重症肌无力[3]。本文患者除胸腺瘤外,有反复肺部感染及指(趾)甲真菌感染、HBV、疱疹病毒感染等机会性感染,同时合并粒细胞缺乏、贫血和血小板减少,临床表现复杂。Good综合征合并粒细胞缺乏的病例罕见[5]。本文患者粒细胞缺乏与粒细胞成熟受阻有关,而粒细胞缺乏进一步导致患者感染风险增加、治疗难度提高,在补充Ig的同时,使用重组人粒细胞刺激因子升白细胞治疗,因此后期临床随访需严密监测粒细胞数量变化。患者免疫缺陷,曾有胆红素升高,贫血需与自身免疫性贫血、溶血性贫血相鉴别。患者铁蛋白升高,贫血逐渐由正细胞性贫血转为小细胞低色素性贫血,骨髓检查未见病态造血,提示其存在慢性病性贫血。患者有慢性乙型肝炎,病毒高度复制,经抗HBV、抗感染治疗后,肝功能、凝血功能和肝纤维化指标改善,血小板升至正常,考虑其血小板减少与感染有关,肝脏肿大与当时病毒感染急性期有关,可排除肝硬化及继发性脾功能亢进。对于胸腺瘤患者,如果合并感染尤其是反复机会性感染或自身免疫性疾病的临床表现时,需警惕Good综合征,完善Ig及T、B淋巴细胞等检查,早期诊断和治疗[6]。而本例患者同时合并粒细胞缺乏、贫血及血小板降低,明确诊断和治疗更为困难。
目前对是否行胸腺瘤切除观点不一,部分学者认为胸腺瘤切除可避免局部浸润,缓解压迫[6]。本患者曾出现呼吸困难,外院胸腺瘤术后缓解,因此我们认为对有压迫症状的患者进行胸腺瘤切除是必要的。Good综合征患者最主要的死亡原因是感染,目前认为定期的Ig替代治疗是提高患者免疫力和预防感染的有效方法[7]。有学者建议每4周检测Ig水平,对于IgG水平不足4 g/L以及免疫反应实验中抗体生成不足者,及时补充Ig[7]。本例患者无B淋巴细胞产生,更需要定期监测及补充Ig,预防机会性感染。此外,近年来有学者尝试利用粪便微生物群移植治疗Good综合征患者的临床效果满意,期望将来更多的临床研究证实此治疗方法的安全性和有效性[8]。
目前Good综合征的发病机制尚未明了。有报道Good综合征患者携带TACI基因突变[9]。另有报道检测到BAFF-R基因突变[10]。然而他们均未阐明TACI、BAFF-R基因突变与B淋巴细胞功能缺陷及Good综合征的关系。本报道中我们发现KIT与ARID1A基因突变。根据文献报道,KIT对骨髓淋巴细胞,尤其是B细胞的发育有重要作用[11]。另有学者报道,ARID1A与免疫检查点调节、T淋巴细胞发育有关[12-13]。但是KIT与ARID1A基因突变是否导致淋巴细胞的发育缺陷从而参与Good综合征的发病过程,需要未来更為深入的基础研究。
参 考 文 献
[1] Lee A Y S, Chockalingam G. Good syndrome: immunodeficiency associated with thymoma. Intern Med J, 2018, 48(7):891-892.
[2] Paranavitane S, Handagala S, De Silva R, Chang T. Thymoma complicated with myasthenia gravis and Good syndrome - a therapeutic conundrum: a case report. J Med Case Rep, 2019, 13(1):348.
[3] Dong J P, Gao W, Teng G G, Tian Y, Wang H H. Characteristics of Goods syndrome in China: a systematic review. Chin Med J (Engl), 2017, 130(13):1604-1609.
[4] Narahari N K, Gongati P K, Kakarla B, Nizami M I, Boddula R P, Sattavarapu L R. Thymoma-associated immunodeficiency: a diagnostic challenge for the clinician. Asian Cardiovasc Thorac Ann, 2017, 25(2):146-149.
[5] Uy K, Levin E, Mroz P, Li F, Shah S. A rare complication of thymoma: pure white cell aplasia in Goods syndrome. Case Rep Hematol, 2019, 2019:1024670.
[6] Guevara-Hoyer K, Fuentes-Antrás J, Calatayud Gastardi J, Sánchez-Ramón S. Immunodeficiency and thymoma in Good syndrome: two sides of the same coin. Immunol Lett, 2021, 231:11-17.
[7] Multani A, Gomez C A, Montoya J G. Prevention of infectious diseases in patients with Good syndrome. Curr Opin Infect Dis, 2018, 31(4):267-277.
[8] Jagessar S A R, Long C, Cui B, Zhang F. Improvement of Goods syndrome by fecal microbiota transplantation: the first case report. J Int Med Res, 2019, 47(7):3408-3415.
[9] Margraf R L, Coonrod E M, Durtschi J D, Augustine N H, Voelkerding K V, Hill H R, Kumánovics A. TACI mutation p.Lys154Ter identified in Good Syndrome. Clin Immunol, 2013, 146(1):10-12.
[10] Lougaris V, Vitali M, Baronio M, Tampella G, Plebani A. BAFF-R mutations in Goods syndrome. Clin Immunol, 2014, 153(1):91-93.
[11] Petkau G, Turner M. Signalling circuits that direct early B-cell development. Biochem J, 2019 , 476(5):769-778.
[12] Shen J, Ju Z, Zhao W, Wang L, Peng Y, Ge Z, Nagel Z D, Zou J, Wang C, Kapoor P, Ma X, Ma D, Liang J, Song S, Liu J, Samson L D, Ajani J A, Li G M, Liang H, Shen X, Mills G B, Peng G. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat Med, 2018, 24(5):556-562.
[13] Astori A, Tingvall-Gustafsson J, Kuruvilla J, Coyaud E, Laurent E M N, Sunnerhagen M, ?hsberg J, Ungerb?ck J, Strid T, Sigvardsson M, Raught B, Somasundaram R. ARID1a associates with lymphoid-restricted transcription factors and has an essential role in T cell development. J Immunol, 2020, 205(5):1419-1432.
(收稿日期:2021-03-24)
(本文編辑:林燕薇)
