北极新奥尔松地区可溶性无机氮盐对根际土壤细菌群落多样性的影响
2021-10-18王瑜韩文冰朱倩蔺立栋王能飞张波涛
王瑜 韩文冰 朱倩 蔺立栋 王能飞 张波涛
(1 青岛大学化学化工学院,山东 青岛 266071; 2 自然资源部第一海洋研究所,山东 青岛 266061)
提要 氮是植物生长的主要营养限制因子,为了探索北极近岸地区植物根际土壤细菌群落多样性受氮元素的影响作用,采用16S rDNA 扩增子测序分析了2014—2016年新奥尔松地区(Ny-Ålesund,Arctic)的3 种典型植物根际土壤和本底土壤样品。结果显示,可溶性无机氮盐(-N,-N,-N)与3 种植物根际土壤整体细菌群落呈显著相关性(P < 0.05)。Nitrosomonadaceae_uncultured (P < 0.05,r2 = -0.77)和 Subgroup 6_norank (P < 0.01,r2 = -0.87)是北极柳根际土壤的核心优势类群,与-N 密切相关。Subgroup 6_norank(P < 0.01,r2 = -0.92)和JG34-KF-361_norank (P < 0.05,r2 = 0.73)与珠芽蓼根际土壤的-N 显著相关。抬头地杨梅根际土壤中的Acidimicrobiales_norank (P < 0.05,r2 = -0.74)和Anaerolineaceae_uncultured (P < 0.01,r2 = 0.93)与-N 有明显的相关性。研究表明,可溶性无机氮盐对北极植物根际土壤细菌群落多样性尤其是核心菌群的变化起到重要作用。
0 引言
当前,全球环境面临着一个重大挑战—— 气候变化迅速。北极气候正在迅速变化,增加了诸如二氧化碳、甲烷和一氧化二氮等温室气体更快释放的可能性[1-2]。Walker 等[3]对苔原生态系统进行变暖实验发现,气温升高使禾本科植物和落叶灌木数量增加,而苔藓和地衣数量减少。气候变化引起的生态环境变动正在逐渐影响陆地生物多样性的分布,这是生物地球化学过程的关键驱动力[4]。微生物在极地陆地环境中占据主导地位,在驱动地球大部分氮循环的生物地球化学过程中发挥着重要作用[5-6]。在生物多样性方面,土壤微生物多样性是植物群落动态的重要调控因子,也是陆地生态系统中植物多样性和生产力的重要驱动力[7],并参与了土壤中大部分养分的转化[8]。Chong 等[9-10]研究南极不同地区土壤微生物的分布格局,指出微生物多样性的特殊性取决于环境的具体理化参数、植被的存在和海拔因素。在很大程度上,环境变化引起的土壤微生物群落结构的改变在土壤理化因子的动态变化中有所体现[11]。因此,通过分析土壤中微生物群落的多样性和结构,可以进一步评估全球变暖对北极生态系统的生物多样性和稳定性的影响机制[12]。
北极的植被类型、生物丰富度和降水量均高于南极,并且北极受全球变暖影响的程度是地球上其他地区的2~4 倍。中国黄河站周围多苔原和湿地,生长的低等植物主要为苔藓和地衣,高等植物比较少,如发草及少数廖科植物,是研究高纬度北极土壤微生物的理想场所[13]。植物和土壤微生物是北极陆地生态系统的两个重要组成部分,它们之间的相互作用是连接地上和地下生态系统的重要纽带[14-15]。北极因其寒冷干燥的恶劣环境,很少能有生长季节超过40~50 天的植物生存,当地占主导地位的生产者主要是垫形植物、莲座丛和簇状禾本科植物[16]。除莎草以外的禾本科植物是极地半荒漠景观的主要植物群落,维管植物覆盖约10%~20%,隐花植物约覆盖40%~70%[17]。
根际被认为是植物的第二基因组,是研究根系、土壤和微生物相互作用的一个热点。越来越多的研究证实,根际微生物的多样性和组成受植物物种[18-19]、土壤类型[20]和根系分泌物的影响[21],其中根系分泌物通过改变土壤的理化性质的方式调节附近的微生物群落[22-23]。20 世纪末,研究报道土壤微生物种群可以对植物根部附近有机养分的释放做出反应,北极地区的植物覆盖区土壤具有更高的微生物多样性[24]。Avery 等[25]的研究结果还表示环境紫外线辐射水平通过影响根分泌物的质量或数量间接影响南极发草的根际微生物多样性。Fierer 和Jackson[26]的研究表明,土壤pH值对微生物多样性分布具有显著性影响。Lemanceau 等[27]和Richardson 等[28]证实土壤中营养物质(如铁、磷酸盐等)的缺乏也会影响微生物群落组成和结构。Glanville 等[29]发现北极苔原生态系统温度和含水量的变化对土壤微生物群落多样性和结构同样具有控制作用。Bueno de Mesquita 等[30]指出,与苔藓覆盖的土壤相比,非植被覆盖的极地土壤的微生物群更少。
而根际微生物进行的生物化学过程也影响着土壤理化特性,两者之间存在双向反馈作用。最新研究发现有植被覆盖的区域,土壤的矿化活动更为活跃[31]。Kumar 等[32]研究北极先锋植物Oxyria digyna(L.) Hill 和Saxifraga oppositifoliaL.的根际土壤细菌多样性和功能,表示根际菌群对氧化应激和特定抗生素的耐受能力与根际群落种类有关。Given 等[33]通过实验证实北极高山植物Oxyria digyna(L.) Hill 的野生根际土壤中含有丰富的磷酸盐增溶和重氮细菌类群。Park 等[34]证实温度升高引起的永久冻土融化和早期融雪增加了土壤含水率,并导致微生物活动增加,故而促进浅层土壤植物有效氮的增加,高水平的土壤氮素又会促进次年夏天灌木的生长。此外,Liu 等[35]从新奥尔松地区的Saxifraga oppositifolia根际土壤中分离到的Pontibacter arcticussp.nov.与重氮菌具有高度相似性。
鉴于目前有关北极植物根际土壤细菌多样性具体受哪些土壤理化因子的影响以及根际细菌群落中哪些优势核心属主导着整体细菌群落结构变化的研究文献较少,我们重点分析了北极3 种典型植物根际土壤理化性质与细菌多样性之间的年际变化及两者之间的相关性。本研究于2014—2016年采集36 个北极新奥尔松地区黄河站附近的北极柳(Salix arctica)、珠芽蓼(Bistorta viviparum)、抬头地杨梅(Luzula confuse)以及无植被覆盖的本底土壤样品,通过对所采集的土壤样品进行16S rDNA 高通量测序,并结合相应的9 种土壤理化性质进行数据分析。结果表明: 3 种根际土壤的主要优势属丰度和可溶性无机氮盐含量具有明显的年际变化特征,且存在显著相关性(P< 0.05)。本研究的目的是将北极土壤的理化参数与根际微生物数据进行关联,从而获得更多北极地区根际细菌群落多样性的年际变化信息,从而进一步探索微生物资源的应用价值,为预测全球气候变化和构建北极土壤生物多样性和生态系统多样性提供有力的数据支持。
1 材料与方法
1.1 研究地点和土壤取样
实验土壤样品采集于北极新奥尔松地区(78° 40'N~79°00'N,11°20' E~12°30'E)。 该地区的植被主要以苔原和湿地为主,生长的低等植物主要是苔藓和地衣,而高等植物较少,例如Oxyria digyna(L.) Hill,Bistorta viviparumL.,Dryas octopetalaL.,Salix arcticaPall.,Luzula confusaLindeb.,Saxifraga nivalisL.等。本研究所选的的3种植物(北极柳、珠芽蓼、抬头地杨梅)是北极新奥尔松地区黄河站附近的常见植物物种。
北极柳(Salix arctica)是一种分布在北极的匍匐落叶灌木,被称为后来的冰川地区定居者,是斯瓦尔巴群岛(Svalbard)裸露山脊的优势物种,能形成外生菌根。它的高度通常低于10 cm,呈垫形。最新研究指出由于其较高的叶片光合作用能力,北极柳在碳固存中发挥着关键效力[36]。珠芽蓼(Bistorta vivipara)是一种环极地的多年生草本植物,生长在北极和高山苔原上[37],是少数能形成外生菌根的草本植物之一,几乎完全通过珠芽无性繁殖[38]。抬头地杨梅(Luzula confuse)是北极高地维管植物类群中最常见的种类之一,常生长于潮湿的生境中,是广泛分布在黏性土壤到沙质土壤中的环极物种[39],其低根冠比、低叶温、高水势和低叶阻的特性是对北极低温和强辐射做出的适应性改变,主要以内生菌根为主[40]。
连续3年选取北极地区3 种优势植物(Salix arctica、Bistorta vivipara、Luzula confusa)根际土壤样品作为研究对象,编号分别为Sar、Bvi 和Lco。选择无植被覆盖土壤作为对照组,编号BG。这些样本来自中国第6 次、第7 次和第8 次北极科学考察。采集样品的方法[41]是在距植物根附近土壤的表面土壤2~5 cm 范围内,用无菌铲刮取约50 g 土壤样品,直接放入无菌塑料取样袋,一式三份,平行样本之间间隔1 m 并呈三角形分布(图1a)。样品在中国的北极考察站黄河站-20℃的冰箱中暂存,然后乘飞机送到国内实验室。在飞行过程中,样本保存在装有冷冻冰袋的恒温箱中。在国内实验室,土壤样品在-80 ℃冷冻,直到核酸提取[42]。共36 个土壤样品。

图1 a)新奥尔松地区土壤样品采集; b)采样站位全景图Fig.1.a) Soil sample collection in Ny-Ålesund (Arctic); b) panorama of sample collection stations
1.2 土壤样品理化性质
实验测量了总共9 种土壤理化性质,即pH值、含水率(MC)、有机碳(TOC,Total Organic Carbon)、有机氮(TON,Total Organic Nitrogen)、-N、-Si、-N、-P 和-N(图2)。MC 的测定为湿土(每个样品10 g)在105 ℃条件下烘干至恒重[43]后的重量失重。称取2 g 土壤加入5 mL 去离子水(现用现制并超声处理),涡旋振荡1 min,静止30 min 后用pH 计(PHS-3C,上海仪电科学仪器股份有限公司,上海,中国)测量pH。为测量其他7 项理化性质,每个样本称取8 g 土壤置于60 mm 培养皿中并编号,放入冻干机中冻干48 h,并用玛瑙研钵研磨成粉末。有机碳和有机氮按照以下程序进行测定。称取 0.1~ 0.2 g 土壤样品,用10%的盐酸处理,随后用去离子水多次冲洗以完全去除盐酸,置于50 ℃干燥后用元素分析仪(EA3000,Euro Vector SpA,Milan,Italy)[44]测定,含量以百分比表示。取2 g 土壤到50 mL 离心管中,按1∶10(g·mL-1)的比例加20 mL去离子水。每4 h 震荡一次,处理48 h 后,用营养自动分析仪(QuAAtro,SEAL,Germany)按相对标准偏差< 5%[45]测定其他五项营养盐(-N,-Si,-N,-P,-N)含量[46-47]。
1.3 DNA 提取和PCR 扩增
从36 个土壤样本(3年 × 4 种土壤类型 × 3 个平行样本:Salix arctica+Luzula confusa+Bistorta vivipara+ 空白)中提取基因组DNA[48]。使用MO BIO Power Soil DNA Isolation Kit(MO BIO Laboratories,San Diego,CA,USA),按照厂家说明从0.25 g 土壤样品中提取,用琼脂糖凝胶检测DNA提取物的纯度和浓度,选择合格的样品[42]进行后续实验。使用引物 806R(5′-GGACTACNNGGG TATCTAAT-3′)和 341F(5′-CCTAYGGGRBGCAS CAG-3′)扩增16S rRNA 基因的V3-V4 区。所有PCR 反应在25 μL 的体系内进行,包括15 μL Phusion R 高保真PCR Master Mix(New England Biolabs,Ipswich,MA,USA),0.2 mmol·L-1正向和反向引物,10 ng 模板DNA。PCR 产物与等体积的上样缓冲液(1×,含SYBR green)混合,装入2%琼脂糖凝胶中检测。选择分子量在400~450 bp 之间的样品,使用Gene JET 凝胶萃取试剂盒(Thermo Scientific,Waltham,MA,USA)进行纯化[47,49]。扩增子文库用 TruSeq®DNA PCR-Free Sample preparation Kit(Illumina,USA) 制备 ,用QuantiFluor™- ST (Promega,USA)进行定量[50]。
1.4 测序和数据分析
纯化后的扩增产物在北京诺禾致源有限公司的Illumina MiSeq 平台上进行测序。使用微生物生态学定量分析(QIIME,version 1.7.0)工作流程对序列进行质量过滤、嵌合体检查、划分可操作分类单元(OTUs)[51]。原始的16S rRNA 基因序列读数用QIIME 软件过滤以去除引物和barcodes,然后用FLASH 软件合并[48,52],利用Uparse 软件对所有的有效序列按相似度97%(Identity)进行聚类成OTUs,并对每个OTUs 的代表序列进行物种注释,物种数据库选择SSU rRNA,得到细菌的物种分类学信息。对数据均一化处理后,使用R 语言和QIIME 计算土壤微生物的α 和β-多样性[53]。通过α-物种多样性和β-物种多样性结果评估微生物多样性的丰富度和均匀度。α-多样性分析包括Chao1、香农指数(Shannon)、辛普森指数(Simpson)和Good’s coverage。β-多样性分析能反映出样本之间的多样性距离关系,而且还可以反映生物群落之间的分化程度,从而用于确定土壤理化性质与细菌群落多样性之间的相关性[48]。采用线性判别效应量(LEfSe)方法对不同采样位点间差异显著的细菌类群进行鉴别(LDA score > 4)。 在相关性分析之前,先对3年土壤理化性质进行常规统计——变异系数(Coefficient of Variation,CV),消除测量尺度和量纲的影响,以表示每个样本理化性质的总体变异性和两组差异明显的数据的离散程度[54]。
标准差与平均数的比值称为变异系数,由下式计算得到:

根据去趋势对应分析(DCA)结果,第一轴长度小于3.0,我们选择冗余分析(RDA)揭示土壤理化性质与微生物类群之间的关系,然后通过蒙特卡洛置换试验对其进行检验。为确定三种根际土壤微生物群落的核心微生物群,采用R 统计包 v3.0.1 的“加权基因共表达网络分析”(WGCNA)进行共表达网络的构建和对比分析[55],然后使用Cytoscape_v3.7.2 构建4 种土壤样品的OTUs 网络图。对4 种根际土壤的核心优势属和9 个理化因子进行Pearson 相关分析(P< 0.05)。原始读取数据存入NCBI 序列读取存档(SRA)数据库(登录号: SRP270629)。
2 结果
2.1 土壤理化性质
2014—2016年BG 平均pH 值为弱碱性,时间变异性低(CVpH= 8.30%)。与同年非根际土壤相比,3 种根际土壤9 个环境因子的变异系数在2014年为2.25%~134.99%,2015年为4.15%~ 119.64%,2016年为1.83%~141.42%。Nielsen 和Bouma[56]对变异系数提出了以下3 种不同类型的土壤特性变异: 0~15%变异不大; 10%~100%为中度变异性; 100%表示高变异性。表1 中的土壤理化性质变异数据显示,除土壤pH 值外,其他因子CV多数超过20%。特别是-N、-N、-N 和-P这4 个因子的变异系数甚至超过100%,表现出高度变异。

表1 36 个土壤样品9 种土壤理化性质的变异系数Table 1.Coefficients of variation of nine geochemical properties of 36 soil samples
2.2 细菌多样性分析和群落组成
从36 个土壤样品中总共获得2025313 条原始序列,经软件质量过滤后得到1520090 条序列。36 个土壤样本的有效序列按97%的序列相似水平共聚类得到5569 个OTUs,每个样本中OTUs平均超过1347 个,其中Lco14_2(站点Lco14 的第2 个样本)包含的OTUs 最多(2690),Sar14_3 包含的OTUs 最少(1244)。样品中OTUs 的覆盖估计值在97%~99%之间,说明测序结果能更全面地反映调查区域环境微生物群落的真实情况。Chao1指数的变化范围为1535.56~3126.72,其中指数最大的3 个站分别为Lco14、Sar16 和Lco16,物种数量最多(表2)。香农(Shannon)指数在Lco14、Sar16 和Sar15 中较高,说明微生物多样性较高,种群丰富,分布均匀。Lco15、BG16、Sar14 值较小,群落结构相对简单。根据物种累积箱线图(species accumulation boxplot,图3a)显示,随着样本个数增加,曲线上升趋势趋于平缓,并结合稀释曲线(图 3b)观察得到样品量足够且测序数据量合理,结果具有代表性,由此反映出 36个土壤样本中物种十分丰富,物种分布均匀,且逐渐趋于5600 多个物种。
在分类学上,我们可以清楚地看到,4 种土壤样本在门水平上有不同的聚类时间模式。首先,每年的3 个Lco 样本不仅形成了独特的集群,也彼此分开(图4)。第二,尽管相对不那么明显,另一根际和BG 样本彼此在同一个年密切相关,图4中清晰地显示出聚集在一起的倾向。第三,2014年的三个Lco 样品与2016年的两个根际样品聚在一起,相关性较强。
在门水平上,无论是根际土壤还是非根际土壤,均以变形菌门(Proteobacteria,27.16%~67.13%,平均46.15%)、放线菌门(Actinobacteria,7.30%~19.74%,平均12.78%)、酸杆菌门(Acidobacteria,4.33%~ 18.11%,平均11.53%)、芽单胞菌门(Gemmatimonadetes,3.39%~21.71%,平均 9.24%)、拟杆菌门(Bacteroidetes,1.68%~17.64%,平均8.50%)占优势(图4)。

图2 2014—2016年土壤理化因子浓度直方图Fig.2.Histogram of concentration of soil geochemical factors from 2014 to 2016

表2 36 个土壤样品的多样性测序数据汇总Table 2.Summary data for diversity sequencing data from 36 samples

图3 a)物种累积箱线图; b)基于16S rRNA 基因V3-V4 的稀释曲线Fig.3.a) Species accumulation curves; b) rarefaction curves based on V3-V4 of 16S rDNA gene
在属水平上,我们发现在所有土壤样品中,Gemmatimonadaceae_uncultured (Gemmatimonadetes,Gemmatimonadetes,Gemmatimonadales,2.86%~ 15.23%),Sphingomonas(0.67%~19.42%),Nitrosomonadaceae_uncultured (Proteobacteria,Betaproteobacteria,Nitrosomonadales,1.19%~7.16%)和Subgroup 6_norank (Acidobacteria,0.77%~8.22%)为优势类群(排名前 4),亚优势类群为 MNG7_ norank(Proteobacteria,Alphaproteobacteria,Rhizobiales,0.56%~3.99%),JG34-KF-161_norank (Proteobacteria,Alphaproteobacteria,Sphingomonadales,0.06%~6.16%),JG34-KF-361_norank (Proteobacteria,Alphaproteobacteria,Rhizobiales,0.11%~4.91%)和RB41(0.07%~7.45%)。
除此之外,我们还比较了土壤理化因子对土壤细菌群落多样性的影响。根据LEfSe 结果,36个样本中有50 个类群的LDA score(线性判别分析)大于 4(显著性检验的截止值)(图 5)。 Sar16 和 Sar14 的分类群最多,分别为 20%和18%。这两个地点的分类单元较其他地点更为丰富,包括变形菌门、拟杆菌门、厚壁菌门和酸杆菌门。
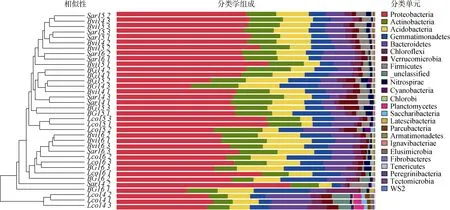
图4 36 个土壤样品中细菌群落组成在门水平的相对丰度Fig.4.Relative abundance of bacterial community compositions at Phylum levels in 36 soil samples
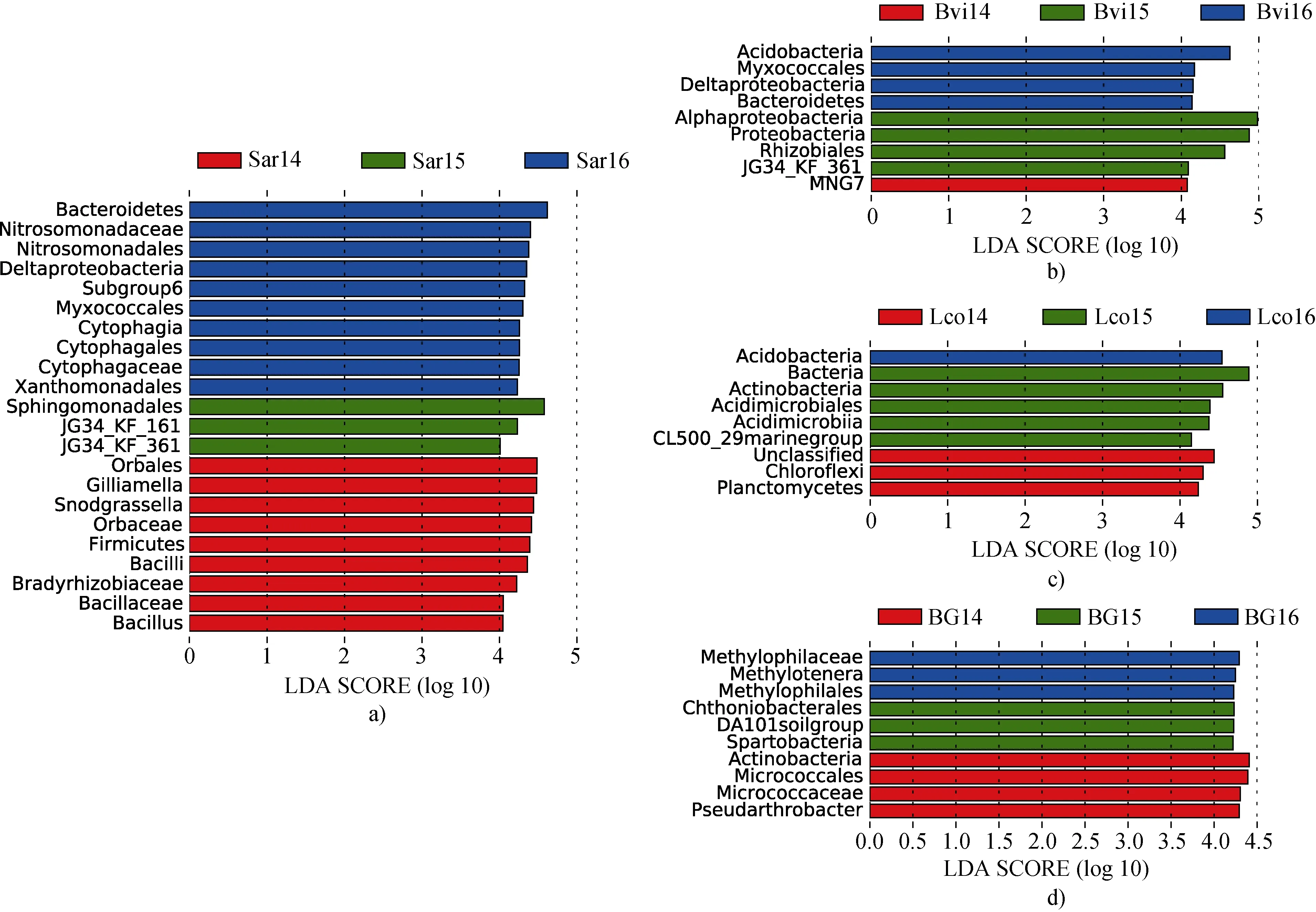
图5 2014—2016年四种土壤样品的LEfSe 分析(默认预设值为4.0,P < 0.05 视为显著).a)北极柳根际土壤; b)珠芽蓼根际土壤; c)抬头地杨梅根际土壤; d)本底土壤Fig.5.LEfSe analyses of four soil samples from 2014 to 2016 (default preset value = 4.0,P-values < 0.05 considered significant).a) Salix arctica rhizosphere soil; b) Bistorta vivipara rhizosphere soil; c) Luzula confusa rhizosphere soil; d) background soil
2.3 土壤理化性质与细菌群落的相关性分析
为了探究理化因素在根际土壤整体细菌群落多样性中是否随时间的变化而发挥不同的作用,我们进行了RDA 分析(图6)和蒙特卡洛置换检验(表3)。

表3 4 种土壤的环境因子与细菌群落组成关系的蒙特卡洛置换检验Table 3.A Monte Carlo permutation test of relationship between environmental factors and bacterial community composition of four soils
如图6 所示,每个根际土壤样品的3 个平行样品相互簇聚,不同根际样品相互分离,时间变化明显。首先,我们发现在4 种土壤样品中,除Sar 样品外,相比其他参数,-Si(r2= 0.95,P< 0.01)与细菌群落总体组成相关性强。同时,2016年除Lco 外,-Si 对其他3 种土壤样品的影响更大。其次,Sar 样品的-N(r2= 0.86,P< 0.01)较其他3 种土壤样品与细菌群落总体组成存在显著相关性。第三,2015年Bvi 样品细菌群落结构受-N 影响最大(r2= 0.95,P< 0.01)。RDA 结果显示,3年的Lco 样品与-N 也有一定的相关性。从BG 的RDA 图可以看出,在无植物土壤中,-N、-N、-N 与细菌群落组成的相关性低于其他6 个土壤理化因子。显然,-N、-N 和-N 对影响根际土壤细菌群落的多样性和组成起主要作用。此外,36 个土壤样本的RDA 结果(图7)表明,3 种根际土壤整体细菌群落与本底土壤发生明显分离。9 项理化性质的蒙特卡洛置换检验(表4)表明,-N(r2= 0.3378,P< 0.01)、TOC(r2= 0.3414,P< 0.01)、含水率(r2= 0.2883,P< 0.01)、pH(r2= 0.2157,P< 0.05)、TON(r2= 0.2118,P< 0.05)对36 个样本的整体细菌群落产生显著影响,而-N(r2= 0.1428,P= 0.079)相关性较低,-N 和-P 与细菌群落没有显著相关性。

图6 基于距离的冗余分析.a)北极柳根际土壤; b)珠芽蓼根际土壤; c)抬头地杨梅根际土壤; d)本底土壤Fig.6.Distance-based redundancy analysis.a) Salix arcticarhizosphere soil; b) Bistorta vivipara rhizosphere soil; c) Luzula confuse rhizosphere soil; d) background soil

图7 基于距离的冗余分析显示2014—2016年36 个土壤样品中细菌群落与环境因子之间的相关性.箭头代表测量的土壤理化因子Fig.7.Distance-based redundancy analysis revealed correlations between bacterial communities and environmental factors in 36 samples from 2014 to 2016.The arrows represent geochemical factors measured

表4 36 个样本的环境因子与整体细菌群落组成关系的蒙特卡洛置换检验Table 4.A Monte Carlo permutation test of relationship between environmental factors and overall bacterial community composition in 36 samples
我们利用加权基因共表达网络分析(WGCNA)的结果(前200 个权重值)来确定这些复杂OTUs网络中最有效的OTU,并找到每个根际的核心类群(图8)。不同植被的根际土壤具有相同的核心类群和其特有的核心类群,但它们的相对丰度各不相同。我们发现四个站点土壤中的共同核心类群为Subgroup 6_norank (Acidobacteria)。Sar 和Bvi共有的核心类群为Gemmatimonadaceae_uncultured
(Gemmatimonadetes,Gemmatimonadetes,Gemmatimonadales),Bacillus。Sar 和Lco 共有的核心类群为Nitrosomonadaceae_uncultured (Proteobacteria,Betaproteobacteria,Nitrosomonadales)。此外,这4 种土壤的OTUs 网络都有其特有的核心类群: Sar 站位有 Cytophagaceae_uncultured (Bacteroidetes,Cytophagia,Cytophagales); Bvi 有Oryzihumus,H16,JG34-KF-361_norank (Proteobacteria,Alphaproteobacteria,Rhizobiales) 和Sphingomonas;Lco有S0134 terrestrial group_norank (Gemmatimonadetes),RB41,Anaerolineaceae_uncultured(Chloroflexi,Anaerolineae,Anaerolineales),Pedobacter,Massilia,Acidimicrobiales_norank (Actinobacteria,Acidimicrobiia,Acidimicrobiales)和Xanthomonadaceae_uncultured (Proteobacteria,Gammaproteobacteria,Xanthomonadales)。DA101 soil group_norank (Verrucomicrobia,Spartobacteria,Chthoniobacterales),Methylotenera和Gemmatimonas是BG 特有的核心类群。
为进一步探讨根际土壤核心优势类群和土壤理化因子的年际变化,确定它们之间的联系。我们将LEfSe 分析得到的差异显著的类群(图5)和网络分析得到的核心类群与9 项理化因子共同进行Pearson 相关分析(Pearson 相关系数见表5)。结合各位点优势类群,我们发现在Sar 中,处于核心位置、丰度较高且差异显著的类群包括Subgroup 6_norank (Acidobacteria),Cytophagaceae_uncultured (Bacteroidetes,Cytophagia,Cytophagales),Nitrosomonadaceae_uncultured(Proteobacteria,Betaproteobacteria,Nitrosomonadales)和Bacillus。Bvi 中存在 Subgroup 6_norank (Acidobacteria),JG34-KF-361_norank (Proteobacteria,Alphaproteobacteria,Rhizobiales),Oryzihumus,H16 和Sphingomonas。以及,Lco 中 Acidimicrobiales_ norank (Actinobacteria,Acidimicrobiia,Acidimicrobiales),RB41,Anaerolineaceae_uncultured (Chloroflexi,Anaerolineae,Anaerolineales)。而BG的优势核心类群为 DA101 soil group_norank (Verrucomicrobia,Spartobacteria,Chthoniobacterales)和Methylotenera。显然,植物类型的差异导致了土壤核心优势菌群的变化。这与蒙特卡洛置换检验的结果一致。Pearson 分析结果表明,优势核心类群与土壤中可溶性无机氮盐具有较强的功能对应性。
3 讨论


图8 36 个土壤样品中细菌群落加权基因共表达网络图.a)北极柳根际土壤; b)珠芽蓼根际土壤; c)抬头地杨梅根际土壤; d)本底土壤.颜色越深,尺寸越大,相关性越强; 相反的比较弱Fig.8.Network diagram of weighted gene co-expression of bacterial communities in 36 soil samples.a) Salix arctica rhizosphere soil; b) Bistorta vivipara rhizosphere soil; c) Luzula confuse rhizosphere soil; d) background soil.The darker the color,the larger the size,the stronger the correlation.The opposite is weaker

表5 土壤理化性质与根际土壤/本底土优势细菌类群丰度的相关系数Table 5.Pearson correlation coefficients of relative abundance of the dominant bacterial groups in rhizosphere soils and bulk soils with soil properties

续表5
在分类学上,我们发现4 个根际土壤的细菌群落具有不同的时间聚类模式。在分类学水平上的统计分析表明,3 种根际土壤的优势菌群组成相似,但在不同根际土壤样品中发现了不同的差异。如细菌群落多样性的变化和特定功能微生物组的存在。根际土壤核心优势类群丰度变化与土壤理化因子之间是否存在交互作用呢?
据基于距离的冗余分析和蒙特卡洛置换检验结果可知,除了pH 值和MC 外,新奥尔松地区植物根际土壤的7 种土壤理化性质与根际细菌群落多样性和组成显著相关。其中,3 种可溶性无机氮盐与细菌群落多样性的相关性最高。在Sar 位点,-N (r2= 0.86,P< 0.01)相关性最高。在Bvi 位点,-N (r2= 0.93,P< 0.01)的相关性最大,而在Lco 位点,-N (r2= 0.86,P< 0.01)的相关性最大。本底土壤中,-Si (r2= 0.80,P< 0.01)的相关性最强,其他理化因素次之。与其他可溶性营养盐相比,根际土壤中可溶性无机氮盐与细菌群落整体多样性和组成显著相关。Van Der Heijden 等[57]研究发现,根分泌物间接介导了有益微生物和病原微生物的相互作用,对根际土壤细菌的多样性和组成起着关键作用。对比 3年的数据发现,3 种根际土壤的-N、-N和-N 理化性质的变异系数呈年变化趋势。分析了两组数据的相关性,发现3 种根际土壤中核心优势类群的丰度与3 种可溶性无机氮盐的变化是一致的。
由于近年来北极环境的变化,土壤中的氮循环发生了显著的变化。受相关功能菌丰度变化的影响,土壤的硝化、反硝化和厌氧氨氧化过程均受到不同程度的促进和抑制。WGCNA 结果显示,Nitrosomonadaceae_uncultured(Proteobacteria,Betaproteobacteria,Nitrosomonadales)既是北极柳根际土壤的优势菌类群,也是其核心菌群。通过相关分析,发现该属与-N 呈现显著负相关(P< 0.01)。此外,核心优势类群Subgroup 6_norank(Acidobacteria)的相对丰度的年际变化和Nitrosomonadaceae_uncultured (Proteobacteria,Betaproteobacteria,Nitrosomonadales)是一样的。同时发现,该类群与根际土壤中-N 和-N 的相关性高于本底土壤。Nitrosomonadaceae_uncultured (Proteobacteria,Betaproteobacteria,Nitrosomonadales)和Subgroup 6_norank (Acidobacteria)的年际变化趋势相同,而Bacillus的年际变化趋势与前两者相反。2014—2015年,-N 和-N 浓度呈协同作用,两者对亚硝酸盐呈现拮抗作用。基于此,我们认为-N 和-N 的浓度影响了Subgroup 6_norank (Acidobacteria)和Nitrosomonadaceae_uncultured (Proteobacteria,Betaproteobacteria,Nitrosomonadales)的相对丰度,-N 浓度显著影响芽孢杆菌的相对分布。前人研究发现,Nitrosomonadaceae_uncultured(Proteobacteria,Betaproteobacteria,Nitrosomonadales)是一种促进土壤硝化作用、争夺土壤-N 的土壤硝化剂[56,58]。Subgroup 6_norank 属于酸性细菌(Acidobacteria),并对土壤氮循环中的-N 和-N 浓度有降低作用[59]。Beneduzi 等[60]在巴西水稻根际土壤中发现芽孢杆菌具有固氮和促进植物生长活性的作用。最近研究指出,在北极新奥尔松地区先锋植物Oxyria digyna和Saxifraga oppositifolia的根际土壤中发现潜在固氮细菌β-变形杆菌和蓝细菌主导土壤细菌群落的多样性和组成[61]。因此,在北极柳根际土壤中,3 种营养盐与3 种优势核心类群之间相互制约、相互影响,共同促进土壤的硝化作用和固氮作用的发生,从而促进了落叶灌木的生长。
珠芽蓼和北极柳均为维管植物,但两个根际的优势核心类群不同。Subgroup 6_norank(Acidobacteria)在珠芽蓼根际土壤中仍占主导地位。有趣的是,优势核心属JG34-KF-361_norank (Proteobacteria,Alphaproteobacteria,Rhizobiales)在珠芽蓼根际土壤中的相对丰度年际变化大于Subgroup 6_norank (Acidobacteria),且与-N 浓度高度相关(P< 0.05)。JG34-KF-361_norank(Proteobacteria,Alphaproteobacteria,Rhizobiales)属于α-变形杆菌纲的根瘤菌目。Delgado 等[62]发现,在硝酸盐的参与下,苜蓿根瘤发生了根瘤菌的反硝化作用。由此我们可以推断,在珠芽蓼根际土壤中,珠芽蓼根与 JG34-KF-361_norank (Proteobacteria,Alphaproteobacteria,Rhizobiales)之间也发生了类似的反硝化过程,后者消耗土壤中的硝酸盐。
Acidimicrobiales_norank (Actinobacteria,Acidimicrobiia,Acidimicrobiales)是Lco 的核心类群,与-N 呈强而显著的相关性(r= -0.74,P< 0.01)。Acidimicrobiaceae sp.strain A6 (A6)来自放线杆菌门,最近被鉴定为一种可进行厌氧氨氧化偶联铁还原(Feammox)的微生物[63]。因此,我们推测,同样的生化过程发生在 Acidimicrobiales_norank(Actinobacteria,Acidimicrobiia,Acidimicrobiales)。 Anaerolineaceae_uncultured (Chloroflexi,Anaerolineae,Anaerolineales)与-N 也有很强的显著相关性(r= 0.93,P< 0.01)。Anaerolineaceae_uncultured属绿弯菌门中的厌氧绳菌目,但其在土壤中的厌氧硝化过程目前尚不清楚。然而,我们的数据发现该类群与-N 浓度有非常显著的相关性,因此我们推断该类群在抬头地杨梅根际土壤中进行了反硝化作用,但具体的作用机制有待于进一步的研究验证。同时,Lco 位点中唯一的门——浮霉菌门(厌氧氨氧化)和蓝藻细菌(固氮),在北极植物根际土壤的氮循环中也发挥着至关重要的作用[64]。Humbert 等[64]发现浮霉菌门参与氨的厌氧氧化产生N2并释放到大气中。相关研究结果表明,与Livingston 岛采集的样品相比,北极地壳样品中蓝藻细菌丰富度更高[65]。一些蓝细菌形成异质囊并具有固定大气氮的能力[66]。然后,蓝藻细菌通过提供固定氮(通过固定氮菌株)在生物降解中发挥着同样重要的间接作用。
本研究发现的核心菌类群主要参与土壤氮循环的主要生化过程,与3 种无机氮盐(-N,-N,-N)的变化显著相关。植物根系对微生物驱动的土壤养分循环有很强的影响[57]。硝化和反硝化是微生物参与大气氮循环的主要途径之一,也是陆地生态系统氮流失的主要原因[57,67]。由于核心优势菌在土壤细菌群落中占有主要地位,其变化对整个细菌群落的多样性和结构变化起着主导作用。综上所述,我们认为,北极根际土壤细菌群落的多样性和组成与3 种土壤可溶性营养盐的浓度变化(如-N,-N,-N)不可分割,且两者的相互作用为北极生态系统抵御气候变化提供了主要保障。
