The utility of endogenous glycochenodeoxycholate-3-sulfate and 4 β-hydroxycholesterol to evaluate the hepatic disposition of atorvastatin in rats
2021-10-18MingynYunjieHunWngGuoqingZhngJinyeDiAn
Mingyn Yunjie Hun Wng Guoqing Zhng Jinye Di -An
a School of Pharmacy, Lanzhou University, Lanzhou 730000, China
b Department of Pharmacy, the First Hospital of Lanzhou University, Lanzhou 730000, China
ABSTRACT The liver is an important organ for drugs disposition,and thus how to accurately evaluate hepatic clearance is essential for proper drug dosing.However,there are many limitations in drug dosage adjustment based on liver function and pharmacogenomic testing.In this study,we evaluated the ability of endogenous glycochenodeoxycholate-3-sulfate (GCDCA-S) and 4 β-hydroxycholesterol (4 β-HC) plasma levels to evaluate organic anion-transporting polypeptide (Oatps)-mediated hepatic uptake and Cyp3a-meidated metabolism of atorvastatin (ATV) in rats.The concentration of ATV and its metabolites,2-OH ATV and 4-OH ATV,was markedly increased after a single injection of rifampicin(RIF),an inhibitor of Oatps.Concurrently,plasma GCDCA-S levels were also elevated.After a single injection of the Cyp3a inhibitor ketoconazole (KTZ),plasma ATV concentrations were significantly increased and 2-OH ATV concentrations were decreased,consistent with the metabolism of ATV by Cyp3a.However,plasma 4 β-HC was not affected by KTZ treatment despite it being a Cyp3a metabolite of cholesterol.After repeated oral administration of RIF,plasma concentrations of ATV,2-OH ATV and 4-OH ATV were markedly increased and the hepatic uptake ratio of ATV and GCDCA-S was decreased.KTZ did not affect plasma concentrations of ATV,2-OH ATV and 4-OH ATV,but significantly decreased the metabolic ratio of total and 4-OH ATV.However,the plasma level and hepatic metabolism of 4 β-HC were not changed by KTZ.The inhibition of hepatic uptake of GCDCA-S by RIF was fully reversed after a 7-d washout of RIF.Plasma concentration and hepatic uptake ratio of GCDCA-S were correlated with the plasma level and hepatic uptake of ATV in rats with ANIT-induced liver injury,respectively.These results demonstrate that plasma GCDCA-S is a sensitive probe for the assessment of Oatps-mediated hepatic uptake of ATV.However,Cyp3a-mediated metabolism of ATV was not predicted by plasma 4 β-HC levels in rats.
Keywords:Atorvastatin Hepatic disposition Glycochenodeoxycholate-3-sulfate 4 β-hydroxycholesterol Oatp Cyp3a
1.Introduction
The liver is an important organ for drugs disposition.However,physiological changes including illness can affect its function and drug disposition,warranting drug dosage adjustment.It is thus very important to be able to accurately measure and predict hepatic drug disposition.Biological markers of the liver function (ALT,AST and ALP) are widely used as markers for drug dosage adjustments.In fact,measures of liver function do not necessarily reflect the changes in the disposition of drugs in the liver [1] .For example,in a cohort of patients with mild to moderate chronic liver disease,the oral clearance of debrisoquine was not significantly affected[2] .Although pharmacogenomic testing is considered to be an important mean to determine precise drug therapy,the genotypes and content of some Cyps have only limited effects on metabolic activities [3],and making drug adjustments based on genotype are not suited for all patients [4,5].We propose a new approach to evaluate hepatic drug disposition by measuring endogenous substrates levels.
Drug disposition in the liver includes substrate uptake from the circulation,intracellular metabolism,and efflux into the bile or blood.Indeed,there are many transporters and metabolic enzymes expressed in hepatocytes that mediate transmembrane transport and metabolism of drugs[6,7].Uptake transporters belonging to the superfamily of solute carriers are located on the basolateral membrane of hepatocytes and include Oatps,Ntcp,Oat2 and Oct1,which mediate the active uptake of bile salts,organic anionic and cationic compounds.Efflux transporter P-gp,Mrp2,Bcrp and Bsep are expressed on the canalicular membrane,where they play an important role in the hepatobiliary excretion of xenobiotics and metabolic products.However,Mrp1,Mrp3 and Mrp4 also expressed in the basolateral membrane and move substrates back into the plasma.Metabolizing enzymes,which include phase I metabolizing enzymes and phase II metabolizing enzymes,biotransform lipophilic compounds into water-soluble metabolites that are more readily eliminated from the body.Among the phase I metabolizing enzymes,P450 superfamily plays the most important role and includes Cyp1a2,Cyp2c9/19,Cyp2e1,Cyp2d6 and Cyp3a4 [8] .Phase I metabolites can be conjugated to endogenous ligands by phase II enzymes (e.g.,Ugts,Sults,Gsts,Nats,etc.) for further transformation into more hydrophilic metabolites [9] .
The human OATP transporters,OATP1B1 and OATP1B3(Oatp1b2 in rodents [10]),are membrane influx transporters that mediated the uptake of endogenous compounds and clinically important drugs from the blood into hepatocytes[11,12].Subsequently,these drugs may be metabolized by phase I/II enzymes.Among these metabolizing enzymes,Cyp3a4 catalyzes the biotransformation of approximately 50% −60% of drugs [13] .Of note,the substrates of Oatps and Cyp3a4 in the liver are mutually overlapping,and include atorvastatin,aliskiren,cerivastatin,repaglinide,nateglinide glibenclamide,lopinavir,simeprevi [14,15].Thus,synergistic actions between Oatps and Cyp3a4 can be important in the hepatic elimination of a diverse range of drugs.How to evaluate the functions of these enzymes and transporters in vivo remains challenging due to the limited detection means for their activity.In recent years,endogenous biomarkers have been proposed for evaluating function of transporters and metabolizing enzymes or for estimating the risk of drugdrug interactions [16,17].Several endogenous substrates of Oatps have been identified in animals or in vitro,including bile acids and their respective conjugates,bilirubin and coproporphyrin I/III (CP I/III) [17] .4β
-Hydroxycholesterol (4β
-HC) [18–21] and 6β
-hydroxycortisol (6β
-HC) [22–25] are formed by the Cyp3a4/5-catalyzed metabolism of cholesterol and cortisol,respectively.Consequently,these compounds may be expected to the endogenous marker for Oatps and Cyp3a.Atorvastatin (ATV) is a widely prescribed hydroxymethylglutaryl-coenzyme A (HMG-CoA) reductase inhibitor that efficiently lowers both cholesterol and triglyceride levels.ATV is subject to cellular membrane uptake by Oatps and is then converted into two major active metabolites,2–hydroxy-atorvastatin (2-OH ATV)and 4–hydroxy-atorvastatin (4-OH ATV) by Cyp3a4 in the liver [26] .Thus,Oatps and Cyp3a4 together determine the hepatic disposition of ATV.The objective of this study was to determine if endogenous substrates for Oatps and Cyp3a can be used to evaluate the hepatic disposition of atorvastatin in rats.In this study,rifampin (RIF) [27–29] and ketoconazole(KTZ) [30,31] were used as an inhibitor of Oatps and Cyp3a4,respectively.
2.Materials and methods
2.1.Materials
Reference materials and pharmaceutical ingredients are shown Table S1.Methanol and acetonitrile were of high performance liquid chromatography (HPLC)-grade (Fisher Scientific,NJ,USA).All other reagents and solvents were of analytical grade and were commercially available.
2.2.Animals
Male Wistar rats aged 6 months weighing 280–350 g,were obtained from the Experimental Animal Center of Lanzhou Institute of Biological Products Co.,Ltd (Lanzhou,China).Rats were housed in plastic cages and maintained at 25 °C under a 12 h alternating light-dark cycle with free access to food and water.All studies were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
2.3.Study design
The first series of experiments was to identify endogenous substrates for Oatps and Cyp3a in rats.In the first experiment,rats were divided into four groups:Control,RIF(30),RIF(60)and RIF(120) group (n
=7 each) and were treated orally with 0.5% CMC–Na,RIF (30 mg/kg),RIF (60 mg/kg) or RIF (120 mg/kg),respectively.Blood and hepatic tissue samples were collected 2 h after a single administration of RIF,and bile acids and their respective conjugates and CP I/III were determined by LC-MS/MS.In the second experiment,rats were divided into four groups:Control,KTZ(20),KTZ(40) and KTZ(80) group(n
=7 each) and were treated orally with 0.5% CMC–Na,KTZ(20 mg/kg),KTZ (40 mg/kg) or KTZ (80 mg/kg) for a single or 7 d consecutively .Blood and hepatic tissue samples were collected at 1 h after the final (or single) KTZ administration,and 4β
-HC and 6β
-HC levels were determined by LC-MS/MS.The second series of experiments investigated the effect of RIF or KTZ on the plasma concentration of ATV and glycochenodeoxycholate-3-sulfate (GCDCA-S) and 4β
-HC.In the first experiment,rats were divided into four groups:Control,RIF,ATV and ATV+RIF group (n
=6 each).The Control and ATV group rats were intravenously administered 50%propylene glycol,and the RIF and ATV+RIF groups were given RIF (30 mg/kg in 50% propylene glycol).The Control and RIF groups were treated orally with 0.5% CMC–Na,and the ATV and ATV+RIF groups were administered ATV (10 mg/kg in 0.5% CMC–Na).In the second experiment,rats were divided into four groups:Control,KTZ,ATV and ATV+KTZ group (n
=6 each).The Control and ATV group rats were intravenously administered 50% propylene glycol,and the KTZ and ATV+KTZ groups were given KTZ (20 mg/kg in 50%propylene glycol).The Control and RIF groups were treated orally with 0.5% CMC–Na,and the ATV and ATV+RIF groups were treated orally with ATV (10 mg/kg in 0.5% CMC–Na).Serial blood samples (0.20 ml) were collected through the right femoral artery at 0,7.5,15,30,45,60,90,120,240 and 360 min.The third series of experiments determined the effect of long-term administration RIF and KTZ on plasma and tissue concentrations of ATV,GCDCA-S and 4β
-HC.Rats were divided into three groups:Control,RIF and KTZ group rats (n
=7 each) were treated orally with 0.5% CMC–Na,RIF (30 mg/kg in 0.5% CMC–Na) or KTZ (20 mg/kg in 0.5% CMC–Na) respectively for 7 d consecutively.All rats were orally administered ATV(10 mg/kg in 0.5% CMC–Na) and blood and tissue samples were collected at 30 min.The fourth series of experiments measured the reversibility of RIF-and KTZ-mediated inhibition of Oatps and Cyp3a.(1) Rats were divided into two groups:Control and RIF group rats (n
=7 each) were treated orally with 0.5%CMC–Na or RIF (30 mg/kg in 0.5% CMC–Na) respectively for 7 d consecutively and then the RIF was allowed to clear for 7 d (2) Control and KTZ group rats were treated orally with 0.5% CMC–Na or KTZ (20 mg/kg in 0.5% CMC–Na) respectively for 7 d consecutively,and then the KTZ was allowed to clear for 7 d,at which times blood and hepatic tissue sample were collected on Day 8 and 14.The fifth series of experiments was to evaluate the influence of liver injure on the hepatic disposition of ATV using the endogenous biomarker of GCDCA-S.A rat model of liver injury was established by oral administration ofα
naphthylisothiocyanate (ANIT).Rats were divided into four groups:Control,ANIT (10),ANIT (30) and ANIT (70) (n
=7 each) and were treated orally with corn oil,ANIT (10 mg/kg),ANIT (30 mg/kg),ANIT (70 mg/kg),respectively.After 48 h,all rats were orally administered ATV (10 mg/kg).Blood and hepatic tissue samples were collected 30 min after a single administration of ATV.ATV and its metabolites and GCDCA-S were determined by LC-MS/MS,AST,ALT and ALP were determined by a fully automated chemistry analyzer(OLYMPLLS AU2700,Olympus Co.,Tokyo,Japan).2.4.Sample preparation and LC-MS/MS analysis
Blood samples were centrifuged at 18 000g
for 10 min.The supernatant plasma layer (100 μl) was mixed with 100 μl of the internal standard solution (IS) and 300 μl methanol for deproteinization.Tissues were weighed and homogenized(0.3 g/500 ml saline),and then centrifuged at 18 000g
for 10 min,after which supernatant (100 μl) was added to 100 μl of the IS with 300 μl methanol.Following vortex mixing and centrifugation at 18,000g
for 10 min,the upper layer was analyzed with by an Agilent 1260 HPLC coupled to an Agilent 6460 Tripe-Quadrupole mass spectrometer equipped with ESI or APCI interface.Quantification was performed using multiple reaction monitoring (MRM) (Table S2).The calibration curves were linear over the concentration range with coefficient (r
) of 0.99 for compounds.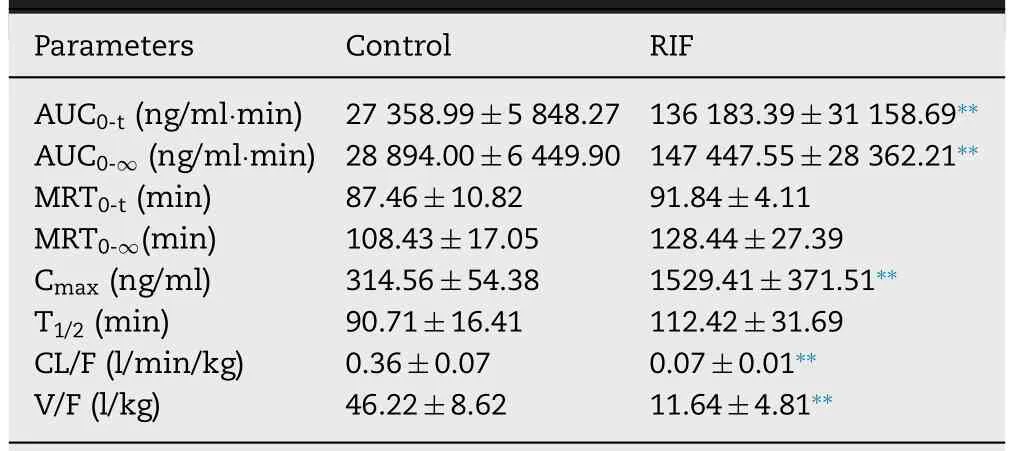
Table 1–Pharmacokinetic parameters of atorvastatin after intravenous administration of RIF in rats.
2.5.Cell uptake and metabolism study
HepaRG cells were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai,China).The cells were grown in RPMI 1640 medium (HyClone,Logan,UT) supplemented with 10% (v/v) fetal bovine serum,100 U/ml of penicillin,and 100 mg/ml of streptomycin at 37 °C in 5% (v/v) COatmosphere.The uptake of ATV and GCDCA-S were measured using ATV and GCDCA-S (10 μM) in the presence or absence of RIF (0.1,0.5,1,5,10 and 50 μM).Intracellular ATV and GCDCAS concentrations were determined.The metabolism of ATV and cholesterol were tested using ATV and GCDCA-S (10 μM)in the presence or absence of KTZ (10 μM),and intracellular 2-OH ATV,4-OH and 4β
-HC concentrations were determined by LC-MS/MS.IC50 was calculated by non-linear regression analysis using GraphPad Prism 5 (GraphPad software,La Jolla,USA),and the calculation equation is as follows:
Where at each inhibitor concentration,the uptake ratio of ATV was expressed as percent of the control (corresponding incubation without inhibitor).X is the inhibitor concentration,and Background is the uptake remaining with maximum inhibition.
2.6.Statistical analysis
Statistical analysis was computed using IBM SPSS Statistics 22 software.Data are expressed as mean ± SD.Statistically significant differences of data from two sets were compared using one-way analysis of variance.In all statistical analyses,P
<
0.05 orP
<
0.01 were considered to indicate a statistically significant result.3.Results and discussion
3.1.Targeted screening for endogenous substrates of OATPS and CYP3A in rats

Fig.1–Plasma concentration (A) and hepatic uptake ratio (B) of BA after a single oral dose of RIF (30,60 and 120 mg/kg).Data are expressed as mean ±SD,n=7,∗P < 0.05;∗∗P < 0.01 compared to the control group.CA,Cholic acid;CDCA,Chenodeoxycholic acid;DCA,Deoxycholic acid;UDCA,Ursodeoxycholic acid;HDCA,Hyodesoxycholic acid;LCA,Lithocholic acid;GCA,Glycocholic acid;GCDCA,Glycochenodeoxycholic acid;GCDCA-S,glycochenodeoxycholate-3-sulfate;GDCA,Glycodeoxycholic acid;GUDCA,Glycoursodeoxycholic acid;TCA,Taurocholic acid;TCDCA,Taurochenodeoxycholic acid;TDCA,Taurodeoxycholic acid;TUDCA,Tauroursodeoxycholic acid;THDCA,Taurohyodeoxycholic acid;TLCA,Taurolithocholic Acid.
The liver makes a major contribution to metabolism and elimination of a variety of drug and directly influences drug exposure levels and therapeutic effectiveness.Thus,the precise assessment of hepatic clearance of drugs in the liver is particularly important in a clinical setting.Jones N.S noted that endogenous biomarkers are becoming important tools to evaluate transporter function,thus providing early assessment for transporter-mediated drug-drug interactions for investigational drugs [32] .Therefore,it may be valuable to estimate or predict the hepatic clearance of drugs by measuring the plasma level of endogenous transporter substrates and their hepatic metabolism.Various substrates of Oatps and Cyp3a have been identified from endogenous metabolites and food-derived compounds.Bile acids and their conjugates and CP I/III have been proposed as endogenous probes for hepatic Oatps [33],and 6β
-HC and 4β
-HC as endogenous probes for Cyp3a [22] .In this study rats were treated with RIF (a specific inhibitor of Oatps [34]) or KTZ(a potent inhibitor of cyp3a [35]).Bile acids and their conjugates and CP I/III,substrates of Oatps,were monitored.As expected,a dose-dependent increase in the plasma and hepatic concentration of RIF was observed over the dose range of 30–120 mg/kg (Fig.S1).The plasma unbound fraction of RIF was 0.265 [29] and so its free concentration was 11.6,22.4 and 26.5 μM at a dose of 30,60 and 120 mg/kg of body weight,respectively.At those concentrations,RIF (ICvalues 0.14–1.7 μM for Oatps in vitro) [29]) could effectively inhibit the hepatic uptake substrates of Oatps,but not affect liver function (Fig.S2).Consistent with previous studies that showed a dose-dependent increase in many bile acids in rat plasma after oral administration of RIF,RIF dose-dependently increased plasma CA (P
<
0.05),CDCA,HDCA (P
<
0.05),GCA,GUDCA,TCDCA,TDCA,THDCA and GCDCA-S (P
<
0.05)levels and decreased the hepatic uptake ratio of CA (P
<
0.05),GCA (P
<
0.05),GUDCA,TCDCA (P
<
0.05),TUDCA,THDCA (P
<
0.05),TLCA (P
<
0.05) and GCDCA-S (P
<
0.05)(Fig.1).Among these metabolites,there is a significant negative correlation between RIF dose and a decrease in the hepatic uptake ratio of CA and GCDCA-S,suggesting that CA and GCDCA-S can be used as endogenous probes of Oatps in rats.GCDCA-S is synthesized from GCDCA in hepatocytes by sulfotransferase and ultimately reabsorbed in the distal small intestine,leading to reuptake into hepatocytes by Oatps(enterohepatic circulation of bile acid) [36] .RIF did not affect the biosynthesis of GCDCA-S from GCDCA in the liver (Fig.S3),suggesting that increase in the plasma concentration of GCDCA-S can be attributed to the inhibition of Oatps by RIF.As shown in Fig.2,RIF did not affect the plasma concentrations of CP I/III and did not inhibit hepatic uptake in our study with rats.Some studies have demonstrated that CP I and CPIII are endogenous substrates for OATP1B in humans [37,38];additionally,Bednarczyk,D showed that CPI is a substrate of OATP1B1 and CPIII is a substrate of OATP1B1 and OATP2B1 [39] .These results suggest that OATPs have different affinities for CPI and CPIII.In rodent liver,the Oatp1a1,Oatp1a4,Oatp1b2,and Oatp2b1 transporters are highly expressed.Although Oatp1b2 in rodent is an orthologs of human OATP1B1 and OATP1B3,there are differences in transport specificity between Oatp1b2 and OATP1B [10,40].This may be the reason for the differing results after administration of RIF in the plasma CP1/III between human and rats.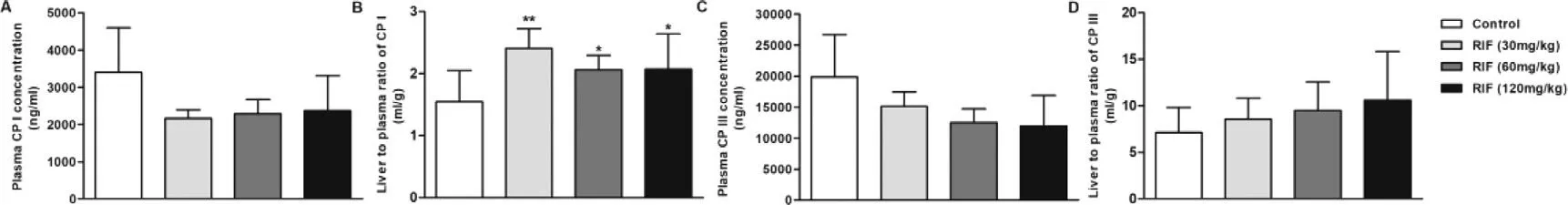
Fig.2–Plasma concentration (A and C) and hepatic uptake ratio (B and D) of coproporphyrin I/III after a single oral dose of RIF (30,60 and 120 mg/kg).Data are expressed as mean ±SD,n=7,∗P < 0.05;∗∗P < 0.01 compared to the control group.
Plasma and hepatic concentrations of KTZ increased in a dose-dependent manner across the dose range of 20–80 mg/kg (Fig.S4).Hepatic concentrations of KTZ were 87.1,119.2 and 160.6 μmol/kg tissue at single oral dose of 20,40 and 80 mg/kg,respectively,and plasma concentration were 23.9,31.4 and 49.6 μM,respectively.KTZ is a potent inhibitor of cyp3a and is often used in vivo to assess the impact of inhibition of Cyp3a on the metabolism of drugs [41] ;its IC50 is 1.06 μM [31] .These results suggest that KTZ levels after a single or multiple-dose can both inhibit the Cyp3amediated substrates metabolism,but not affect liver function(Fig.S2).Hepatic 4β
-HC levels were significantly increased(Fig.S5),and the metabolic ratio of 4β
-HC to cholesterol were not affected significantly by KTZ (Figs.3 A-3 C).Although Cyp3a could be inhibited with a single dose of KTZ,this may not have been sufficient to quantitatively change plasma metabolites levels.Thus,rats were treated with KTZ for 7 d consecutively.Unexpectedly,the plasma concentrations of 4β
-HC were markedly elevated after repeated administration of KTZ,but the metabolic ratio of 4β
-HC to cholesterol showed a dose-dependent decrease (P
>
0.05) (Figs.3 D-3 F),suggesting that 4β
-HC is a poor plasma biomarker for Cyp3a activity in rats.Although many studies have shown that 4β
-HC could be utilized as a Cyp3a activity marker [19,20,42–45],the metabolic ratio of 4β
-HC to cholesterol was not influenced by single or multiple-dose of KTZ in our study.This could be attributed to the fact that the change in plasma 4β
-HC concentration was not sensitive to the inducer and inhibitor due to the long half-life (half-life of~17 d) [46,47].Some studies have proposed that urinary 6β
-HC could be used as a non-invasive marker for Cyp3a induction or inhibition [22,24,25,45,48].At a single administration of KTZ hepatic 6β
-HC levels were dosedependently decreased by KTZ,but plasma levels were not been influenced (Fig.4).After multiple-dose treatment with KTZ the blood concentration of 6β
-HC increased.Previous studies have demonstrated that 6β
-HC underwent uptake by OAT3 in the basolateral membrane of renal tubular epithelial cells and efflux by MATE1,MATE2-K or P-gp in the apical membrane [49] .Nevertheless,KTZ could inhibit the efflux transporters,MATE1,MATE2-K and P-gp [50,51].These results suggest that the increase in plasma 6β
-HC concentration can be related to the inhibited transporter-mediated urinary elimination of 6β
-HC by KTZ,and the decrease in hepatic 6β
-HC levels can be related to the inhibition of Cyp3a activity.However,many studies have shown a stronger preference for 4β
-HC as endogenous marker than 6β
-HC [19,52,53].
Fig.3–Plasma concentration of 4 β-HC (A and D) and cholesterol (B and E) and metabolic ratio of 4 β-HC to cholesterol in the liver (C and F) after a single (A,B and C)and multiple doses (D,E and F) of KTZ (20,40 and 80 mg/kg).Data are expressed as mean ±SD,n=7,∗P <0.05 compared to the control group.
Together with these findings,GCDCA-S and 4β
-HC (Fig.S6) were selected as endogenous substrate of Oatps and Cyp3a,respectively,to evaluate the hepatic disposition of atorvastatin in rats.3.2.Effect of RIF on ATV and GCDCA-S pharmacokinetic
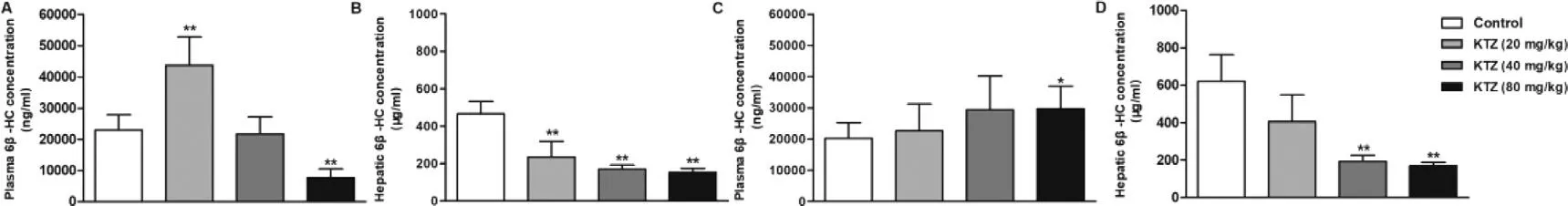
Fig.4–Plasma (A and C) and hepatic (B and D) concentration of 6 β-HC after a single (A and B) or 7 multiple doses (C and D)administration of KTZ (20,40 and 80 mg/kg).Data are expressed as mean ±SD,n=7,∗P < 0.05;∗∗P < 0.01 compared to the control group.
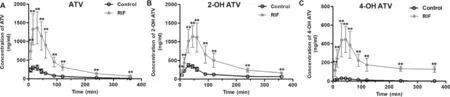
Fig.5–Plasma concentration-time curves of ATV (10 mg/kg,p.o.) (A) and its metabolites,2-OH ATV (B) and 4-OH ATV (C)after intravenous administration of RIF (30 mg/kg).Data are expressed as mean ±SD,n=6,∗P < 0.05;∗∗P < 0.01 compared to the control group.
ATV is taken up into hepatocytes by Oatps and then metabolized to 2-OH ATV and 4-OH ATV by Cyp3a [26] .Decreases in Oatps and Cyp3a activity could result in an elevated blood exposure to ATV,perhaps leading to myopathy.Thus,it is important to evaluate the function of Oatps and Cyp3a for the safe use of statins.In this study,we first evaluated the correlation between the change in GCDCA-S and the hepatic uptake of ATV.The pharmacokinetic parameters of ATV,a probe drug of Oatps,were determined in rats after a single intravenous administration of vehicle or RIF(Fig.5 and Table 1).Plasma concentrations of ATV and its metabolites,2-OH ATV and 4-OH ATV,were markedly higher in rats treated with RIF (30 mg/kg) than in control rats.However,CL/F and V/F of ATV were significantly decreased by RIF.Concurrently,plasma concentrations of GCDCA-S were significantly increased after intravenous RIF administration(Fig.6).In addition,RIF did not affect the plasma concentration of cholesterol and 4β
-HC (Fig.S7).These results suggest that the plasma RIF level (above 5.5 μM,Fig.S8) can inhibit the hepatic uptake of ATV and GCDCA-S.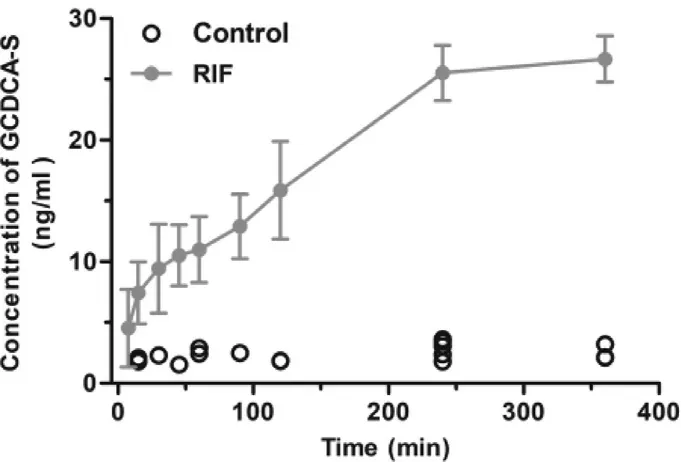
Fig.6–Plasma concentration-time curves of GCDCA-S after intravenous administration of RIF (30 mg/kg) (n=6).
3.3.Effect of KTZ on ATV and 4 β-HC pharmacokinetic
The effect of KTZ on the pharmacokinetic of ATV and its metabolites was also evaluated after a single intravenous administration of KTZ (Fig.7 and Table 2).Plasma concentrations of ATV in rats treated with KTZ were significantly increased compared to control rats.By contrast,its metabolites,2-OH ATV and 4-OH ATV concentrations were significantly decreased at 7.5,15,30 and 60 min after KTZ administration.The pharmacokinetic parameter MRT was significantly increased and CL/F decreased,but V/F was not affected by KTZ,suggesting that KTZ predominantly affects the metabolism of ATV,but not its hepatic uptake.Maeda K et al.demonstrated that the rate-limiting process of ATV in the hepatic elimination of ATV is the hepatic uptake in healthy volunteers [54] .However,consistent with our study,Chang,J.H.,et al.demonstrated that the inhibition of Cyp3a by KTZ could result in an increase in plasma ATV concentration in mice,but the plasma concentration of ATV when Oatps was inhibited by RIF was much higher than that when Cyp3a was inhibited by KTZ [30] .The reason for this difference may be attributed to differing species or the use of different types of inhibitors.In addition,plasma concentrations of GCDCA-S were not changed by KTZ (Fig.S9).The plasma levels of cholesterol and its metabolite 4β
-HC were not affected by KTZ (Fig.8).These results suggest that the KTZ level (above 1.5 μM,Fig.S10) can inhibit ATV metabolism,but not inhibit 4β
-HC formation from cholesterol.
Fig.7–Plasma concentration-time curves of ATV (10 mg/kg,p.o.) (A) and its metabolites,2-OH ATV (B) and 4-OH ATV (C)after intravenous administration of KTZ (20 mg/kg).Data are expressed as mean ±SD,n=6,∗P < 0.05;∗∗P < 0.01 compared to the control group.

Table 2–Pharmacokinetic parameters of atorvastatin after intravenous administration of KTZ in rats.
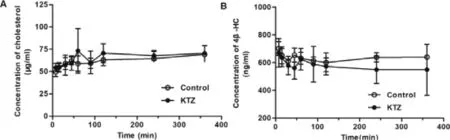
Fig.8–Plasma concentration-time curves of cholesterol (A)and 4 β-HC (B) and after intravenous administration of KTZ(20 mg/kg) (n=6).
3.4.Effect of long-term administration RIF or KTZ on the disposition of ATV, GCDCA-S and 4 β-HC
Considering that Oatps and Cyp3a could not be effectively inhibited after a single dose of RIF or KTZ,respectively,rats were treated orally with RIF or KTZ for 7 d consecutively.As shown in Fig.9,the hepatic uptake of ATV was greatly attenuated by RIF,but minimally impacted by KTZ.Similarly,RIF significantly increased the plasma concentration of GCDCA-S and decreased its hepatic uptake.The metabolic ratio of ATV to 4-OH ATV was decreased by KTZ,but not affected by RIF.On the contrary,plasma levels and the metabolic ratio of 4β
-HC to cholesterol were not changed by KTZ and RIF (Fig.9 and Fig.S11).These results demonstrate that GCDCA-S can be used to evaluate the Oatp-mediated uptake of ATV in the liver of rat,but 4β
-HC levels does not reflect Cyp3a-mediated ATV metabolism.3.5.Effect of long-term administration RIF or KTZ on plasma concentrations of GCDCA-S and 4 β-HC
To investigate whether the inhibitory effects of RIF and KTZ were transient or persistent,rats were given RIF or KTZ for 7 d consecutively,followed by a 7-d washout period.As shown in Fig.10,the plasma concentration of GCDCA-S in RIF-treated rats was higher than that in vehicle-treated rats,and after a 7-d washout period the GCDCA-S level was not significant different between the two groups.The effect of RIF on the hepatic uptake of GCDCA-S was fully reversed after washout of RIF.These results show that GCDCA-S is a sensitive marker for evaluating the change in Oatps function.The effect of KTZ on Cyp3a-mediated cholesterol degradation to 4β
-HC was consistent with the above,and there was no marked difference in plasma concentrations and the metabolic ratio of 4β
-HC after a 7-d washout period.This could be related to the fact that 4β
-HC has a long half-life [42,46,55] and thus its utility as a marker of Cyp3a activity in short-term studies is uncertain due to hysteresis or low intra-individual variation between Cyp3a activity and changes in 4β
-HC.Kasichayanula et al.reported that plasma 4β
-HC concentration continued to increase after 3 d of RIF dosing [42] .In contrast,our study shows that RIF does not influence the metabolic ratio of 4β
-HC to cholesterol after 7 d of RIF treatment (Fig.S12).3.6.Effect of RIF on uptake of ATV and GCDCA-S in HEPARG cells

Fig.9–Effect of RIF (30 mg/kg,p.o.) and KTZ (20 mg/kg,p.o.) after 7 d dosing on ATV uptake (A) and metabolism (B),plasma level (C) and hepatic uptake (E) of GCDCA-S and plasma level (D) and metabolic ratio (F) of 4 β-HC.Data are expressed as mean ±SD,n=7,∗P < 0.05;∗∗P < 0.01 compared to the control group.

Fig.10–Inhibitory effects of RIF and KTZ on hepatic uptake of GCDCA-S and metabolism of 4 β-HC.Plasma level (A) and uptake ratio (B) of GCDCA-S after 7 d of RIF dosing,followed by a 7-d washout period;plasma level (C) and metabolic ratio(D) of 4 β-HC after 7 consecutive days of RIF dosing,followed by a 7-d washout period.Data are expressed as mean ±SD,n=7,∗P < 0.05 ∗∗P < 0.01 compared to the control group.
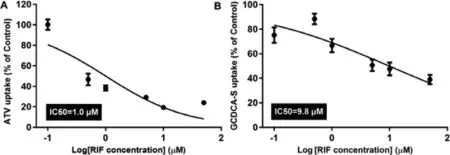
Fig.11–Effect of RIF (0.1,0.5,1,5,10 and 50 μM) on uptake of ATV (10 μM) (A) and GCDCA-S (10 μM) (B) in HepaRG cell(n=5).
The HepaRG cell line is derived from a hepatocellular carcinoma and expresses a large panel of liver-specific genes including several cytochrome P450 enzymes and drug transporters [56,57].Thus,HepaRG cells were selected for their similarity to human primary hepatocytes.Firstly,an MTT (3-(4,5-dimethylthiazol-2-yl) −2,5-diphenyltetrazolium bromide)cell proliferation assay was performed (Fig.S13).The uptake of ATV and GCDCA-S was markedly inhibited by RIF (Fig.11)and IC50 values for RIF with the uptake of ATV and GCDCAS were 1.0 μM and 9.8 μM,respectively.These results suggest that RIF can inhibit the uptake of ATV and GCDCA-S in HepaRG cells.Consistent with this in vivo study,KTZ inhibited the degradation ATV to 2-OH ATV and 4-OH ATV,but did not affect the formation of 4β
-HC (Fig.S14).These results demonstrate that GCDCA-S can be used as a marker for the uptake function of Oatps,but 4β
-HC is an imperfect marker of the metabolic function of Cyp3a.3.7.Correlation analysis between the hepatic disposition of ATV and GCDCA-S
ANIT is commonly used as an indirect hepatotoxic agent to investigate the molecular mechanisms of both acute and chronic cholangiopathy injury [58] .Thus,we investigated the correlation between the hepatic disposition of GCDCA-S and ATV in rats with ANIT-induced liver injury.Serum AST and ALT levels were markedly elevated in rats treated with ANIT (30 and 70 mg/kg) (Fig.12 F),suggesting that liver injury was present after administration of ANIT (30 and 70 mg/kg).Plasma levels of ATV and GCDCA-S were slightly increased (P>
0.05) in the ANIT (30 mg/kg) and ANIT (70 mg/kg) groups,but there was no significant change in 10 mg/kg ANIT-treated rats(P
>
0.05) (Fig.12 A and 12 D).The hepatic uptake ratio of ATV and GCDCA-S was markedly decreased in ANIT (30 mg/kg) and ANIT (70 mg/kg) compared to that of the control group (P
<
0.05) (Fig.12 B and 12 E).The metabolic ratio of ATV to 2-OH ATV or 4-OH ATV in the ANIT group was not significantly different(P
>
0.05) compared with control group (Fig.12 C).Correlation between individual endogenous biomarkers (GCDCA-S,ALT,AST,ALP) and ATV was evaluated.Results showed that the plasma concentration of GCDCA-S was highly correlated with the plasma level of ATV (R=0.94,P
<
0.0001) (Fig.12 G),and hepatic uptake ratio was medially correlated between GCDCAS and ATV (R=0.4671,P
=0.0006) (Fig.12 H).However,there was poor correlation between the serum biomarker of ALT,AST,ALP and plasma concentration of ATV in rats with liver injury (Fig.12 I).Consistent with a previous report by Wang et al.[59],the liver mRNA level of Oatp was decreased in ANIT-induced liver injury (Fig.S15).The decrease of hepatic uptake of GCDCA-S and ATV could be attributed to the down-regulated expression of Oatp in rats treated with ANIT(30 mg/kg) and ANIT (70 mg/kg).These resulted demonstrated that GCDCA-S has a better sensitivity to assess the disposition of ATV in rats with ANIT-induced liver injury.
Fig.12–Correlation analysis between the hepatic disposition of ATV and GCDCA-S in rats with ANIT-induced liver injury.Plasma level of ATV (A) and GCDCA-S (B);hepatic uptake ratio of ATV (D) and GCDCA-S (E);the metabolic ratio of 2-OH ATV and 4-OH ATV (C);serum biomarkers (F);individual correlation analysis between endogenous biomarker (GCDCA-S,ALT,AST,ALP) and ATV (G,H and I) after a single ANIT dosing (10,30,70 mg/kg).Data are expressed as mean ±SD,n=7,∗P <0.05,∗∗P < 0.01 compared to the control group.
In this study,we found that endogenous GCDCA-S instead of biochemical markers (AST,ALT or ALP) can be used to evaluate hepatic ATV uptake.By measuring the serum concentration of it,we can exactly know the activity of hepatic Oatps.As a result,dosage of drugs which were taken up by hepatic Oatps can be adjusted according to plasma GCDCA-S concentration.Because of species differences of OATP between rats and humans,clinical trials and developing PBPK model to quantitatively describe GCDCA-S behavior will be necessary.But most importantly,endogenous biomarkers related to drug transporters or metabolic enzymes may be preferable to accurately evaluate drug disposal in a clinic setting,and as those biomarkers would be directly related to drug disposal,whereas serum biochemical markers (liver function and kidney function) do not directly reflect the distribution,metabolism and excretion of drugs.
4.Conclusions
Our results shows that liver function markers such as AST,ALT and ALP do not always affect drug disposition in the liver,and GCDCA-S can be used as a minimally invasive marker to assess Oatps-mediated hepatic uptake of ATV.However,our study does not support using plasma 4β
-HC levels to reflect Cyp3a-mediated ATV metabolism in rats,perhaps due to the long half-life 4β
-HC in blood.Conflicts of Interest
All the authors declare no conflict of interest.
Acknowledgement
This work was supported by National Natural Science Foundation of China (Grant No.81803611).
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.ajps.2021.03.002 .
杂志排行

Asian Journal of Pharmacentical Sciences的其它文章
- Chondroitin sulfate-mediated albumin corona nanoparticles for the treatment of breast cancer
- Integrated computer-aided formulation design:A case study of andrographolide/ cyclodextrin ternary formulation
- Targeting the resolution pathway of inflammation using Ac2–26 peptide-loaded PEGylated lipid nanoparticles for the remission of rheumatoid arthritis
- The effect of ethanol on the habit and in vitro aerodynamic results of dry powder inhalation formulations containing ciprofloxacin hydrochloride
- Macrophage membrane-mediated targeted drug delivery for treatment of spinal cord injury regardless of the macrophage polarization states
- Size,shape,charge and“stealthy”surface:Carrier properties affect the drug circulation time in vivo