Integrated computer-aided formulation design:A case study of andrographolide/ cyclodextrin ternary formulation
2021-10-18YnWngXiyngnhuiHuHifengDefngOuyng
Yn Wng Xiyng n nhui Hu Hifeng Defng Ouyng
a State Key Laboratory of Quality Research in Chinese Medicine, Institute of Chinese Medical Sciences (ICMS), University of Macau, Macau 999078, China
b Institute of Applied Physics and Materials Engineering, University of Macau, Macau 999078, China
c Tongren Hospital, Shanghai Jiao Tong University School of Medicine, 1111 Xianxia Road, Shanghai 200336, China
d Jiangsu Province Hi-Tech Key Laboratory for Biomedical Research, School of Chemistry and Chemical Engineering, Southeast University,Nanjing 211189, China
ABSTRACT Current formulation development strongly relies on trial-and-error experiments in the laboratory by pharmaceutical scientists,which is time-consuming,high cost and waste materials.This research aims to integrate various computational tools,including machine learning,molecular dynamic simulation and physiologically based absorption modeling(PBAM),to enhance andrographolide (AG)/cyclodextrins (CDs) formulation design.The lightGBM prediction model we built before was utilized to predict AG/CDs inclusion’s binding free energy.AG/ γ-CD inclusion complexes showed the strongest binding affinity,which was experimentally validated by the phase solubility study.The molecular dynamic simulation was used to investigate the inclusion mechanism between AG and γ-CD,which was experimentally characterized by DSC,FTIR and NMR techniques.PBAM was applied to simulate the in vivo behavior of the formulations,which were validated by cell and animal experiments.Cell experiments revealed that the presence of D-α-Tocopherol polyethylene glycol succinate (TPGS) significantly increased the intracellular uptake of AG in MDCKMDR1 cells and the absorptive transport of AG in MDCK-MDR1 monolayers.The relative bioavailability of the AG-CD-TPGS ternary system in rats was increased to 2.6-fold and 1.59-fold compared with crude AG and commercial dropping pills,respectively.In conclusion,this is the first time to integrate various computational tools to develop a new AG-CD-TPGS ternary formulation with significant improvement of aqueous solubility,dissolution rate and bioavailability.The integrated computational tool is a novel and robust methodology to facilitate pharmaceutical formulation design.
Keywords:Integrated computer-aided formulation design Machine learning Molecular dynamic simulation Physiologically based absorption modeling Andrographolide Cyclodextrins
1.Introduction
About 40% of marketed drugs and even 75% of developed drug candidates can be listed in poorly water-soluble drugs based on the Biopharmaceutical Classification system(BCS) [1] .To solve this issue,pharmaceutical scientists have developed multiple soluble formulations.In traditional formulation design,to prepare a product with the desired characteristics,the ratio of drugs and excipients and the process parameters must be optimized [2] .The evaluation of the oral absorption performance of dosage forms is an essential perspective in the pharmaceutical industry,which requires experimental validations [3] .However,the current pharmaceutical formulation design mainly focuses on each pharmaceutical researchers’ trial-and error approaches.Also,the traditional formulation experiment is time-and money-consuming,and it also wastes a massive amount of materials.High-throughput screening technology based on machine learning has been widely used in drug design in the past decade [4],which brought continuous pressure on the productivity of the formulation industry.Thus,to develop accurate and straightforward methods in formulation design becomes more and more critical in the pharmaceutical industries [5] .
With the rapid development of computer science and IT techniques,recently various computational tools are growing to be applied in drug formulation design [6],molecular mechanism exploration [7] andin
vivo
prediction [8],which provide rapid growth potential for the pharmaceutical industry.Machine learning approaches,a subset of artificial intelligence,are computerized algorithms that optimize computer program performance based on the data or previous experience [9] .In our previous works,we had used various machine learning models to predict formulations of oral disintegrating tablets [10],the physical stability of solid dispersions [11] and the complexation performance of cyclodextrins (CDs) inclusions [12] .These machine learning models can provide us a good prediction to benefit the formulation design.To further explore the interaction between drug molecules and excipients,molecular dynamic simulation can mimic the behaviors of complex systems,molecular interactions,aggregations,and diffusions for the complex system under different pressures and temperatures at the atomic level [13] .These molecular details are challenging to be explored by current characterization techniques,such as spectroscopic analysis and X-ray diffraction [14] .By providing threedimensional structures of molecules,it is effortless for scientists to understand the mechanisms between drugs and CDs binary and ternary system,which plays an essential role in drug design and provides a simplification for formulation screening [15] .The ibuprofen-CDs binary systems and lutein-CDs multiple component system were developed by molecular modeling approaches in our previous study.The molecular modeling results showed a high correlation with experimental results and provided a reasonable explanation for the molecular mechanisms [16,17] .
In the preliminary screening of the drug formulation process,the estimation of pharmacokinetic parameters for each formulation beforein
vivo
study is particularly critical.The physiologically based absorption modeling (PBAM) can predict the distribution of APIs in various organs and blood depends on the physiochemical properties of the drug and the route of administration [18],which is essential for the assessment of the bioavailability of the formulation.As the most critical part of formulation research,thein
vivo
study could explore the performance of oral formulation that is absorbed by the gastrointestinal tract and passes through the liver to the systemic circulation.Sun et al.built the intravenous injection (i.v.
) administration PBAM of progesterone to predict the trend of its permeability with different concentration of 2-hydroxypropyl-β
-cyclodextrins(HP-β
-CD).The PBAM proved that the permeability of progesterone could not decrease rapidly with a certain amount of HP-β
-CD [19] .Furthermore,Wang et al.constructed the PBAM bicyclol immediate-release and control-release tablets in the beagle dog and human improving the efficiency of the formulation optimization in the clinical trials [20] .This study aims to integrate various computational tools for CDs formulation design.Andrographolide (AG),a high hydrophobic drug (74 μg/ml at 25 °C in water),was chosen as the model drug.The binding free energy between six kinds of CDs and AG by the lightGBM prediction model we built before [12] .Then,MD simulation investigated the complexation behavior between AG and different CDs.Subsequently,PBAM was used to predict pharmacokinetic parameters of pure AG,AG dropping pill and AG-CD-TPGS ternary formulation.Finally,a series of relative experiments were carried out to validate the whole process simulation result.2.Materials and methods
2.1.Prediction of binding free energy between AG and CDs by machine learning method
The physicochemical properties relative molecular descriptor(molecular weight,complexity,logP
and so on) of AG and 6 kinds of CDs were inputted into the lightGBM prediction model we built before [12] .Then the prediction result of binding free energy was obtained.2.2.Molecular modeling of AG-CD complexation
2.2.1.Model
establishing
for
molecular
modeling
approaches
The structure ofγ
-CD was obtained from Cambridge Crystallographic Data Center,HP-β
-CD,Me-β
-CD and SBEβ
-CD were modified from the structure ofβ
-CD obtained from Cambridge Crystallographic Data Center by Discovery Studio 2016 Client,the position of substituent was referred to previous paper [21,22] The structure of AG was built by Discovery Studio 2016 Client according to Chinese pharmacopeia 2015.All the molecular structures were optimized with a force field by Discovery Studio 2016 Client.2.2.2.Docking
simulation
setting
AutoDock Vina is an open-source software to predict the combination mode of 1:1 ligand-receptor interaction.The AutoDock Tools package and AutoDock Vina was used to build the initial structure of molecular dynamics(MD) simulation [23] .In docking simulation,the semiflexible docking method was adapted to AG,and docking coordinates and the search space center of CDs are shown in Table 1 .AG with different insertions in the direction of CD would be performed in MD simulations as initial structures.
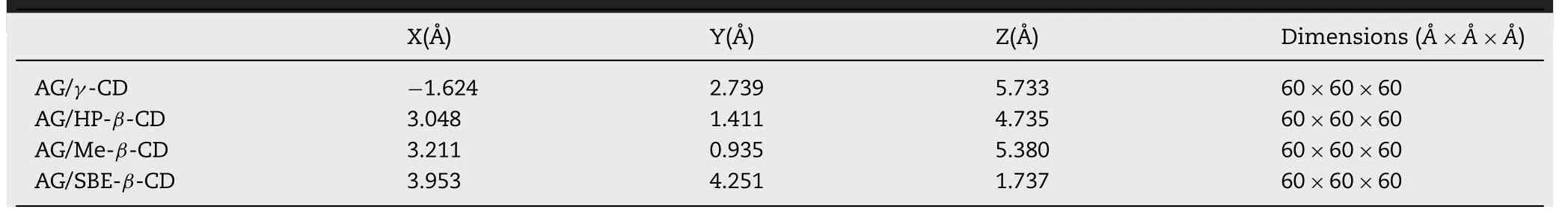
Table 1–Docking Simulation details of different AG-CDs systems.
2.2.3.Molecular
dynamics
simulation
The MD simulations were based on the AMBER 14 software package with the general AMBER force field (gaff) for CDs and AG.The initial structures obtained from docking simulation were immersed in a water box with a 20 °A thickness TIP3P water model with the general AMBER force field(gaff) and the ff14SB force field in LEAP module of Amber Tools 14.In the minimization procedure,the structures of AG and CDs were subjected to 10 000 steps of solvation energy minimization.The whole system was subjected to 20 000 steps of minimization.After minimization,the system was heated to 310 K,and the cutoff distance was 10.0 °A.Then,50 ns MD simulations of 4 systems at 310 K were performed in the solvated system to simulate the molecular mechanisms of AG-CD complexes.Constant pressure periodic boundary with an average pressure of 1 atm was also applied.
2.2.4.MM-PBSA
calculation
The MM-PBSA method was commonly used to calculate the binding free energy of the 1:1 ligand-receptor system [24,25] .And the average interactions between AG and CDs were calculated using the last 500 snapshots structures from the MD trajectory of the system.The binding free energy of AG-CD inclusion complex (Δ
G bind) was calculated by the free energy of complex (Δ
G complex) and the isolated AG (Δ
G AG) and CDs(Δ
G) as the following equations:
Δ
G) was calculated by the enthalpy(Δ
H) and entropy with invariable temperature:
2.3.Physiologically based absorption modeling
2.3.1.Model
structure
The PBAM models in the present work were built on MATLAB 2020a (The MathWorks,Inc.,US) with the SimBiology APP.Modeling for AG was based on the model structure,which simplified the“Generic SimBiology PBPK model”(available on https://www.mathworks.com/matlabcentral-/fileexchange/37752-generic-simbiology-pbpk-model) by replacing the three-compartment model of an original whole-body model with digestion and absorption part unchanged.The resulted model is like the compartmental absorption and transit(CAT) model [26] or the advanced CAT (ACAT) model [27] .CD,dissolved drug,and undissolved drug are considered in the present model.The latter two species can transform into each other by dissolution or precipitation process.The three species transfer through the gastrointestinal tract divided into compartments of the stomach and several intestine segments.The free fraction of the dissolved drug in each segment is calculated in real-time.The free drug can be absorbed.After absorption,the drug undergoes distribution,metabolism and excretion.The systemic disposition is described by a classical three-compartment model.
The dissolution process of the drug is described in Eq.(5) :
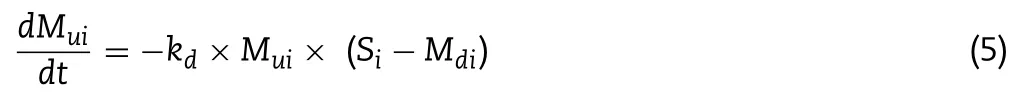
M
for dissolved amount andk
is dissolution rate constant.S
represents the solubility in each segment,and it is calculated according to the CD concentration in the corresponding site and a result of the phase solubility test.In the present work,the precipitation process is also modeled using the same equation in the opposite direction.Transfer of drug from one intestine segment to next one is calculated as:

k
is the transport rate constant,i
indicates each single intestine segment,and C is the concentration of the drug.For duodenum,jejunum and ileum,the transport rate constant is calculated as:
k
ts
is small intestine transport rate constant andL
s
is the length of small intestine whileL
i
is the length of specified small intestine segment.To calculate drug absorption of ternary formulation,a permeability model,which is modified from the work of Dahan et al.[28],has been used.The first step is to define the free fraction of dissolved drug,that is the drug proportion not bound to CD molecule:
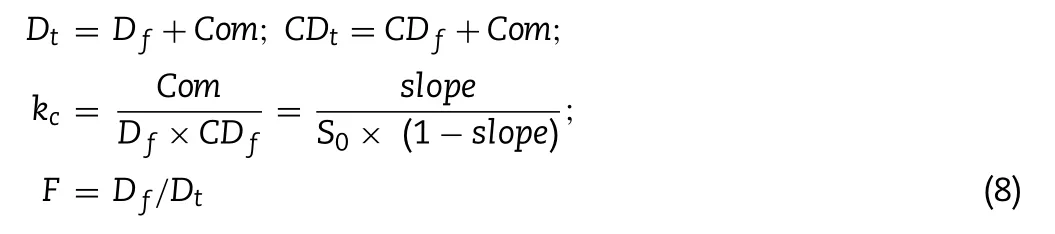
D
t
,D
,CD
t
,CD
andCom
represent the concentration of the total drug,free drug,total CD,free CD and drug-CD complex,respectively;k
c
is the binding constant which depends on the data of aqueous solubility and slope in phase solubility test;S
0
was the aqueous solubility of the drug.As it is assumed that only drug not bound to CD molecule can be absorbed,the free fraction F is incorporated into the calculating method: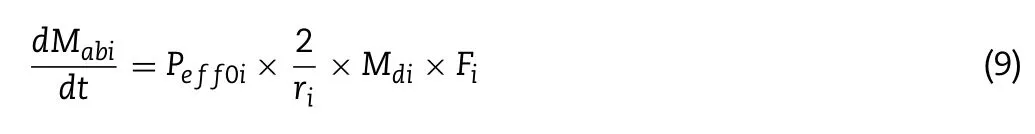
M
is absorbed drug amount;P eff0 is effective permeability in the absence of CD,which is often determined by rat jejunumin
situ
perfusion test;r
is intestine segment radius.The product ofP
andF
can be regarded as permeability in the presence of CD.P
multiplying with2/r
results in the absorption rate constant (k
).The delivery of drugs from the gut to the central compartment is mimicking transport via the portal vein.The equations used here are complicated and known as Rodgers&Rowland method [29,30] .The physiological parameters required by this method share the same value as the original model,while the needed drug properties like logP
,unbound fraction and blood plasma concentration ratio are newly collected.For systemic disposition,the drug is first absorbed into the central compartment.The exchange of drug from central(with index 1) to peripearal_1 (index 2) compartment is modeled as first-order kinetic:

And so does the exchange from central to peripearal_2(index 3):

The metabolism of the drug from the central compartment is like:

At the initial step,the amount of drug and CD in all compartments except for in the stomach is set to 0.The initial CD amount in the stomach is the same as that is used in the formulation.And the initial dissolved and undissolved drug amount in the stomach is calculated according to solubility in the presence of CD in the formulation.
2.3.2.Modeling
for
andrographolide
The simulation’s parameters were mainly maintained unchanged as the original generic PBAM in SimBiology,except for what is mentioned below.The intestine segment radius was extracted from the PK-Sim database(Open System Pharmacology,available on http://www.open-systems-pharmacology.org/).The effective permeability of AG in each intestine segment was referred to the rat in
situ
intestinal perfusion test result [31] .However,permeability values reported in this work are in dimensionless form.Restoring the permeability to its actual value requires,on the other hand,identifying the intestinal radius,and on the other hand,the required diffusion coefficient of AG was calculated as 3.11 ×10cm/s by PK-Sim.Blood plasma ratio and logP
were also determined by PK-Sim.Plasma unbound fraction 0.35 was taken from the literature [32] .AG is considered a neutral compound since its solubility does not change significantly in a pH range [33],so that parameters pK a or pK b were neglected.The PBAM was built based on the dissolution profile with pH 6.8 PBS.Solubility and dissolution rate constant for each formulation was fitted to corresponding dissolution test result in previous work in the first place,except for ternary formulation,where 74 μg/ml [34] was used as aqueous solubility and to calculate real-time solubility.Dosed solid drug and dissolved drug were calculated according to solubility before administration.To make simulation more accurate,transport rate (k
t
) of the colon was fitted to pure AG formulation PK profile to grasp the later part of curve better,and this transport rate has been maintained in next two formulations.After absorption,AG undergoes a systemic disposition process,wherein associated parameters were determined by fitting to 5 mg/kgi.v.
data[35] in advance.The parameters used in AG modeling are listed in Table 2 .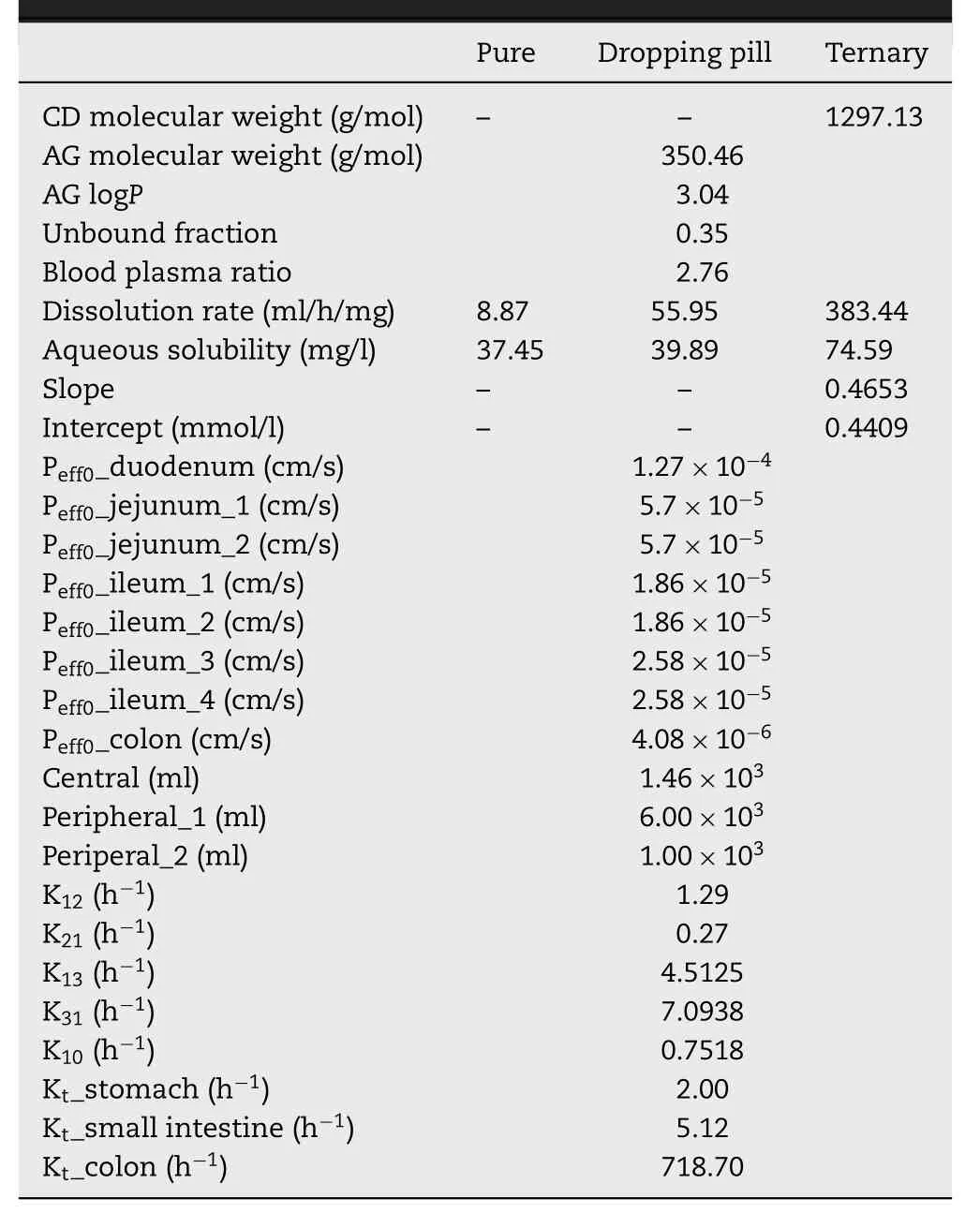
Table 2–Parameters used in AG modeling.
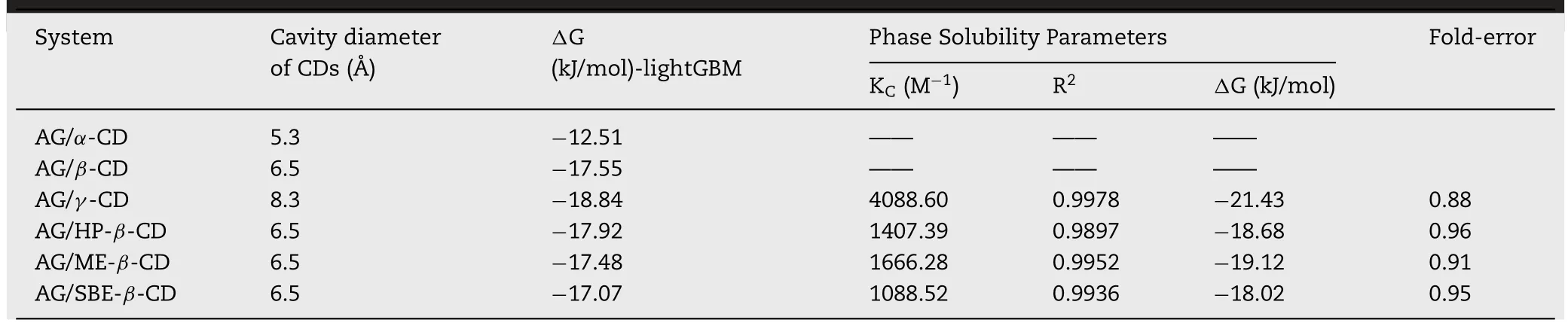
Table 3–The comparison between the predicting and experimental results.
2.4.Experimental validation
2.4.1.Materials
and
reagents
AG was purchased from Yibinyirui Chemical Industry CO.LTD.(Sichuan,China).α
-CD,β
-CD,γ
-CD,HP-β
-CD,Me-β
-CD,D-α
-Tocopherol polyethylene glycol succinate(TPGS) and resveratrol (Internal standard) were purchased from Shanghai Aladdin Bio-Chem Technology.CO.LTD.(Shanghai,China).SBE-β
-CD was purchased from Hubei Tuochukangyuan Chemical Industry.CO.LTD.(Hubei,China).3-(4,5-dimethylthiazol-2-yl) −2,5-diphenyltetrazolium bromide (MTT),dimethyl sulphoxide (DMSO) and poloxamer 407 (P407) were purchased from Sigma-Aldrich (St.Louis,MO).Commercial dripping pills of AG (150 mg),used as a reference,were purchased from TASLY Holding Group.CO.LTD.(Tianjin,China).HPLC grade methanol was purchased from Merck Company (Darmstadt,Germany).All materials for cell culture were obtained from Life Technologies Inc.(GIBICO,USA).Other reagents and solvents for the study were of analytical grade.2.4.2.Phase
solubility
study
The phase solubility investigation was carried out in an aqueous medium as the method reported by Higuchi and Connors previously [36] .Briefly,an excess amountof AG (5 mg) was added to 1 ml aqueous CDs solution with various concentrations ranged from 0 to 15 mM,respectively.The sealed glass vials were shaken on a mechanical shaker at 37 °C for 48 h to reach the state of equilibrium.After that,all the samples were filtered by 0.45 μm cellulose acetate membrane immediately.The filtrate was analyzed by Agilent 1200 series HPLC system (Agilent,USA).The conditions of HPLC were list in supplementary materials.
The apparent stability constant (K) was used to compare the affinity between drugs and different CDs [37,38] .The K value of AG-CDs complexes with 1:1 stoichiometric ratio can be calculated in equation from the phase solubility curve Eq.13 .In addition,the Gibbs free energy (Δ
G) can be obtained by K 1:1 value using Eq.14 .
S
0
was the aqueous solubility of AG,the slope was obtained using linear regression between the molar concentrations of AGversus
CDs in water.
R
was the gas constant,T
was Kelvin temperature and K 1:1 was the apparent stability constant of AG-CDs complexes with a 1:1 stoichiometric ratio.2.4.3.Formulation
screening
of
AG-CDs
binary
system
Low aqueous solubility of AG was the main factor that limited its bioavailability,as mentioned before.In this study,the AGCDs binary systems were designed to enhance the solubility of AG,which consisted of AG and different CDs with the molar ratio of 1:1 or 1:2,respectively.Briefly,the required amountγ
-CD was dissolved in a distilled water to get a clear solution.AG powder was added to the solution in a different molar ratio (1:1 or 1:2) and ground for 2 h until forming the paste.Then the mixture was placed under 50 °C in the vacuum oven to evaporate water.The dried substance was pulverized,sifted through an 80 mesh sieve and stored for the following analysis.The saturated solubility of AG-CDs complexes was detected for the formulation screening by HPLC analysis (the conditions of HPLC were list in supplementary materials).2.4.4.Cell
culture
Madin-Darby canine kidney cells transfected with the multidrug resistance 1 gene (MDCK-MDR1) cells were a kind gift from Prof P Borst (the Netherlands Cancer Institute,Amsterdam,the Netherlands).The use of MDCK-MDR1 was approved by the Institute of Chinese Medical Sciences,University of Macau,for research purposes.MDCK-MDR1 cells were cultured in a dulbecco’s modified eagle medium (DMEM)with antibiotics,including 1% penicillin and streptomycin(P/S),10% heat-inactivated fetal bovine serum (FBS) at 37 °C in an atmosphere of 5% COand 95% relative humidity.
2.4.5.In
vitro
cytotoxicity
assay
The cytotoxicity of MDCK-MDR1 cells was investigated by MTT assay [39] .Cells were seeded on 96-well plates at 1 ×10cells/well in 100 μl of medium and allowed to adhere overnight.After that,cells were treated with indicated concentrations of AG,AG/γ
-CD complexes and AG/γ
-CD complexes with different P-glycoprotein (P-gp) inhibitors for 3 h.Then cells were washed with HBSS and incubated with 100 μl per well of freshly prepared MTT dye for another 4 h.Next,100 μl of DMSO was added into each well to dissolve the formazan crystals.The cell viability was measured based on the absorbance at 530 nm.The experimental concentrations for all tested samples were less than the maximum nontoxic concentrations (cell survival>
90% compared with the control group)2.4.6.Cellular
uptake
The effect of various P-gp inhibitors on restraining drug efflux was assessed by determining cell uptake of AG in MDCK-MDR1 cells.In brief,the MDCK-MDR1 cells were seeded on 6-well plates at 5 ×10cells/well and allowed to adhere overnight.After reaching 80% confluence,the medium was replaced by 100 μM AG in the absence or presence of 200 μMγ
-CD and 1% of four P-gp inhibitors (Tween 80,TPGS,P407 and Cremphor® EL).The same concentration of AG commercial dripping pills was used as the positive control.All the samples were incubated at 37 °C in an atmosphere of 5% CO 2 and 95% relative humidity for 90 min.After that,the cells were washed with ice-cold PBS solution three times and lysed in 100 μl 1% Triton X-100.The content of AG in the lysate was determined by HPLC analysis (the conditions of HPLC were list in supplementary materials).2.4.7.Preparation
of
AG-CD-TPGS
ternary
formulation
AG-CD-TPGS ternary formulation was carried out by the grinding method with the molar ratio of 1:2.Briefly,1% of TPGS was dissolved in a minimum amount of water and sonicated for 30 min.γ
-CD was mixed with the former solution in a mortar,and AG powder was added to the suspension and ground for 2 h until forming the paste.Then the mixture was placed under 50 °C in the vacuum oven to evaporate water.The dried substance was pulverized,sifted through an 80 mesh sieve,and stored for the following analysis.The physical mixture was prepared with the same ratio in the optimal AG-CD-TPGS ternary formulation by uniformly mixing all the components in a sealed container.2.4.8.Characterization
The thermal characters of AG,γ
-CD,TPGS,physical mixture and AG-CD-TPGS ternary system were recorded using a differential scanning calorimetry system (DSC-60A,Shimadzu,Japan).All the samples (3 mg) were weighed accurately and encapsulated in aluminum pans.The temperature range was set from 30 to 250 °C with a heating rate of 10 °C/min under nitrogen.Aluminum oxide was used as a reference standard.The FTIR spectra of AG,γ
-CD,TPGS,formulation physical mixture,and AG-CD-TPGS ternary formulation were recorded with a fourier transform infrared spectrometer (Spectrum 100,PerkinElmer,America) in the scanning range of 4000–400 cmat the resolution of 2 cm.NMR spectroscopy has been previously used to establish inclusion modes and stoichiometries [40,41] .H NMR spectra of AG,γ
-CD and AG-CD-TPGS ternary formulation were recorded at 25 °C with a Bruker ultra-shield 600 plus NMR spectrometer (Bruker,Germany).Deuterium oxide (DO) was used as the solvent.2.4.9.Dissolution
study
The dissolution profile was commonly applied for describing drug releasein
vitro
.In this study,the dissolution study of AG,physical mixture,AG-CD-TPGS ternary formulation and commercial AG dropping pills were performed with a fully automated dissolution tester (DT700,ERWEKA,Germany).According to Ch.P 2015 Apparatus II,the dissolution test method was employed at paddle rotational with a speed of 75 rpm at 37 ±0.5 °C.1000 ml distilled water,0.1 mol/l HCl solution,pH 6.8 phosphate buffer solution (PBS) and 1%sodium dodecyl sulfate solution (SDS) were chosen as the dissolution medium.150 mg AG or other samples equivalent to 150 mg AG were added into 1000 ml dissolution medium,and 5 ml aliquots were withdrawn at predetermined time intervals (5,10,15,20,30,45 and 60 min).The same volume of fresh medium was added immediately to maintain a constant volume.All the withdrawn samples were filtered through 0.45 μm filters for HPLC analysis (the conditions of HPLC were list in supplementary materials).2.4.10.Cell-based
transwell
transport
study
in
MDCK-MDR1 monolayer
A transwell model with MDCK-MDR1 cells was designed to mimic the uptake and efflux process in intestinal cells.Briefly,MDCK-MDR1 cells were cultured onto polycarbonate inserts(22.74 mm ID,0.4 μm pore size,4.06 cmof the growth area,SPL Life Sciences Co.,Korea) with a seeding density of 5 ×10cells/well and cultured for 7 d prior for transport study.The media was changed every other day for the first 4 d and then every day after that.The transepithelial electrical resistance(TEER) at 37 °C was measured with an epithelial voltohmmeter(World Precision Instruments,Inc.,FL,UK) to confirm the integrity of monolayers.Those monolayers with TEER values higher than 140 ohms could be employed for the following study.
The transport experiment was conducted in Hank’s Balanced Stock Solutions (HBSS) at 37 °C.Before the experiment,MDCK-MDR1 cell monolayers were rinsed twice and equilibrated with HBSS buffer for 30 min.The HBSS volumes in the apical (AP) and basolateral (BL) chambers were 1.5 ml and 2.6 ml,respectively.Then 20 μM AG,AG-CD-TPGS ternary formulation or dropping pills containing 20 μM AG in HBSS were loaded onto AP side or BL side to study APBL and BL-AP transfer.1 ml samples were collected at the opposite side at 15,30,45,60,90 and 120 min,and the same volume of blank HBSS was added to the receiver chamber.All the samples were analyzed by HPLC (the conditions of HPLC were list in supplementary materials).Permeability coefficient (P app) and efflux ratio (EfR) were calculated using the following equations reported previously.

Δ
R/Δ
t was the rate of AG appearance in the receiver chamber,S was the surface area of membrane (4.71 cmin this study),and C D0 was the initial concentration of AG in the donor side at time zero.
P
app
(BL-AP)
and P app(AP-BL)
were the permeability values in secretory and absorptive directions,respectively.2.4.11.In
vivo
pharmacokinetic
study
Sprague-Dawley (SD) rats were used for the pharmacokinetic study.The experiment protocols and procedures were approved by Animal Ethical Committee at the Institute of Chinese Medical Sciences,University of Macau,Macao SAR,China (UMARE-029–2016),and were performed with the Guide for the Care and Use of Laboratory Animals.All the rats were fed under standard laboratory conditions (standard laboratory diet,the controlled temperature of 20–22 °C and 50% relative humidity with 12 light/dark cycle).The day before drug administration,an intubation operation was performed for the rats with polyethylene catheters (0.4 mm I.D.,0.8 mm O.D.,Portex Ltd.,Hythe,Kent,England) on the right jugular veins under anesthesia with 10% chloral hydrate.After the operation,the rats were recovered individually and fasted for 12 h with free access to water.
Eighteen male surgically cannulated SD rats (300 ±20 g)were randomly divided into three groups.Each group was administrated with cruder AG,commercial AG dropping pills and AG-CD-TPGS ternary formulation separately.A dose of nearly 12 mg AG (40 mg/kg of body weight) was performedvia gastric gavage.0.3 ml blood samples were manually withdrawn into heparin-coated tubes from the right jugular vein via the cannulated catheter at 0.25,0.5,0.75,1,2,3,4,6,8,10,12 and 24 h after drug administration.The same volume of heparinized saline was injected into rats after each sample collecting to maintain a constant blood volume.All the obtained blood samples were centrifuged at 5 000 rpm for 10 min,and the plasma samples were stored at −80 °C for further analysis.
The plasma sample preparation was performed according to the previous publication with several modifications [42] .Briefly,20 μl internal standard (resveratrol,10 μg/ml) was added into 100 μl plasma samples.After vortex mixing,1 ml ethyl acetate was added into the mixture mentioned above and vortexed for 3 min.Then,samples were centrifuged at 5 000 rpm for 10 min.The clear supernatant was transferred into a plastic centrifuge tube and evaporated under a vacuum drying oven at 45 °C.Finally,the dried residue was reconstituted with 100 μl methanol and for HPLCMS analysis.The HPLC-MS analysis was performed on a Waters e2695 Alliance separations module equipped with a Waters 2998 photo-diode array detector (PDA,Waters,USA)coupled to a mass single-quadrupole detector (QDa,Waters,USA).The conditions of HPLC-MS were list in supplementary materials.The pharmacokinetic parameters were calculated by MonolixSuite 2020R1 (Lixoft,France).
2.4.12.Statistical
analysis
Each experiment was performed in triplicate and was repeated for at least thrice.All the results were expressed as mean ±SD.Statistical significance was evaluated by Student’st
-test using IBM SPSS Statistics 22 software.The differences were considered statistically significant with a value ofP
<
0.05.3.Results and discussion
3.1.Comparison of the prediction and experimental results of binding free energy
3.1.1.The
prediction
result
by
lightGBM
method
The result of the machine learning and phase solubility study was exhibited in Table 3 .As shown in the result of lightGBM,AG/γ
-CD was the most stable system with the lowest binding free energy (−18.84 kJ/mol).Moreover,the binding free energy of AG/β
-CD,AG/HP-β
-CD,AG/ME-β
-CD and AG/SBE-β
-CD were from −17.07 to −17.92 kJ/mol,which was approximate while AG/α
-CD was the unstable inclusion system with the largestΔ
G (−12.51 kJ/mol).The difference ofΔ
G between the prediction value and experimental value ranged from 0.88 to 0.95 fold which showed that this lightGBM model could accurately predict AG-CDs’ binding energy.
Compared with the experimental results,the binding energy of the lightGBM prediction model showed a similar trend.In this model,the prediction of binding energy could be used as the primary screening method before phase solubility experiments.
3.1.2.Phase
solubility
study
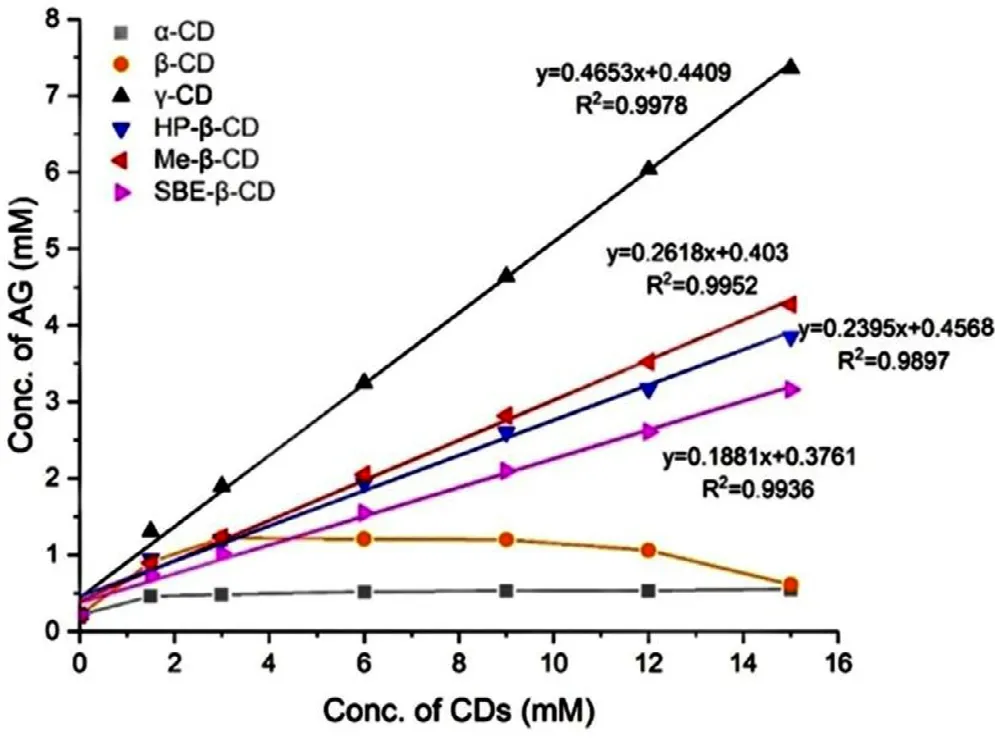
Fig.1–Phase solubility diagram of AG with different CDs in distilled water at 37 ±0.5 °C.
The phase solubility diagram of AG with different CDs was shown in Fig.1 .The typical HPLC chromatograms of AG were shown in Fig.S1.Considering in this diagram,the solubility of AG increased linearly due to the formation of inclusion complex between AG withγ
-CD,HP-β
-CD,Me-β
-CD and SBEβ
-CD,individually.It was clear that the solubility diagram of AG in the presence of these four CDs indicated the Atype and formed a 1:1 complex [44] .The solubility of AG increased with the concentration ofβ
-CD increasing initially,while it remained stable after the concentration ofβ
-CD reached 3 mM and decreased with the further addition ofβ
-CD.It may be attributed to the low aqueous solubility of the AG/β
-CD complex,which belonged to the BS type and reported in previous research.The solubility of AG was not enhanced significantly with the attendance ofα
-CD,which might due to the smallest inner diameter compared with other CDs.The calculation results of K C,RandΔ
G value were shown in Table 3 .The stability constant of AG/γ
-CD was significantly higher than the other three systems with an order ofγ
-CD(4088.6M)>
Me-β
-CD (1666.28M)>
HP-β
-CD (1407.39M)>
SBE-β
-CD (1088.52M).The higher KC suggested a more stable complex had been formed between AG with CDs.All the negative binding free energy showed spontaneous and exothermic processes for the formation of AG-CDs complex.3.1.3.Formulation
screening
of
AG-CDs
binary
system
Since theα
-CD andβ
-CD did not significantly enhance AG’s solubility,the other four types of CDs were considered in the formulation screening.AG-CDs complexs with the ratio of 1:1 and 1:2 were prepared by the grinding method.The saturated solubility of different formulations was shown in Fig.2 .AG exhibited very low saturated solubility (74.59 μg/ml) in distilled water.Compared with pure AG,all the formulations enhanced AG solubility.Besides,AG-CDs complex with a ratio of 1:2 showed more significant solubility improvement than those with a ratio of 1:1.Among the AG-CDs systems,the AG/γ
-CD complex in the ratio of 1:2 exhibited the highest AG solubility and reached 2990.2 μg/ml,which increased approximately 40 folds than pure AG.Therefore,the AG/γ
-CD binary system with the ratio of 1:2 was regarded as the optimal formulation for the enhancement of AG solubility.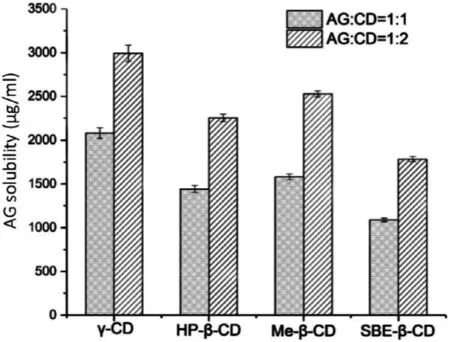
Fig.2–AG saturated solubility of different formulations in distilled water at 37 ±0.5 °C.
3.2.The molecular mechanism of AG-CDs
3.2.1.Molecular
modeling
of
AG-CDs
binary
system
The final structure of the AG-CDs binary system predicted by MD simulation was shown in Fig.3 .According to different inclusion types,two possible structures of AG-CDs complex were exhibited,and the calculation of binding free energy for each reasonable structure by MMPB-SA method was listed in Table 4 .The possible structure of AG-CDs binary system with the lowest binding free energy would be considered the most stable structure.According to the MD simulation predicting results,it seemed that the AG-CDs binary system could form both of two possible inclusion types complex except for the Me-β
-CD,which showed the lactone ring in the CD cavity was not accepted.Also,based on the calculation results of binding free energy,the most stable structure of the AG/γ
-CD complex was the lactone ring resisted intoγ
-CD inner cavity,while the others showed the decalin ring in the CDs cavity.This might be attributed to the different diameters of the inner cavity betweenγ
-CD and otherβ
-CD derivatives.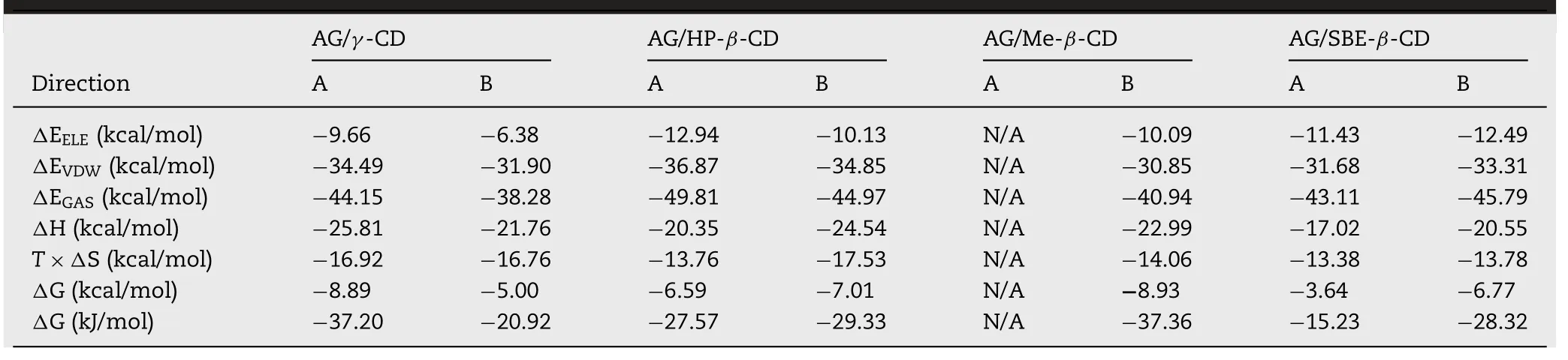
Table 4–Calculation of binding free energy by MMPB-SA for AG-CDs binary system.
3.2.2.Characterization
analysis

Fig.3–Final structure of AG-CDs binary system with different directions predicted by MD simulation.(A) The lactone ring in CD cavity and (B) the decalin ring in CD cavity.
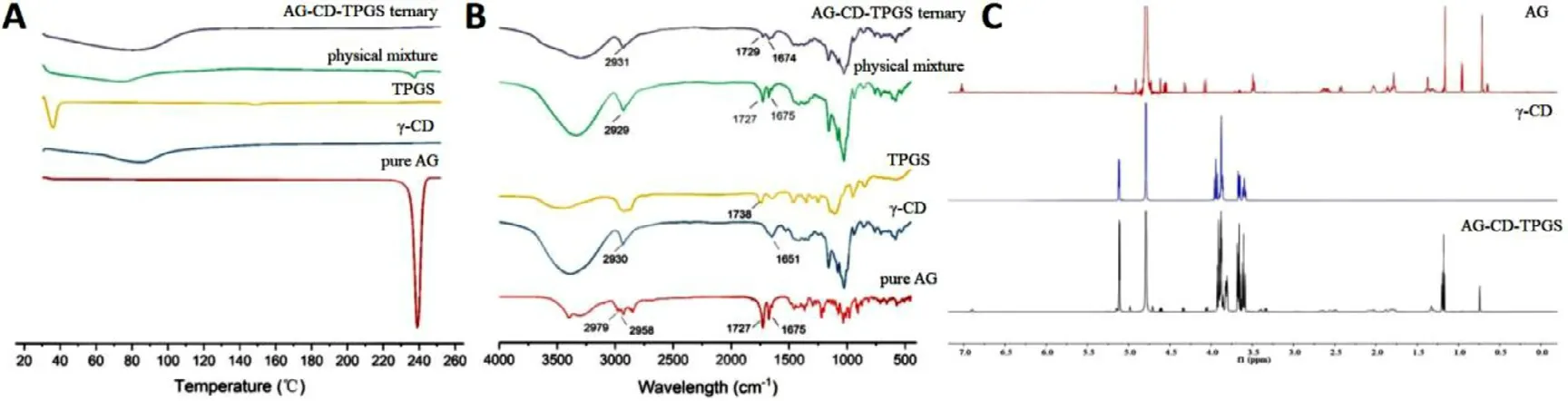
Fig.4–The characterization patterns for (A) DSC thermograms for AG-CD-TPGS ternary formulation,physical mixture,TPGS,γ-CD and pure AG;B FTIR spectra of AG-CD-TPGS ternary fo rmulation,physical mixture,TPGS,γ-CD and pure AG;C 1 H NMR spectra of pure AG,γ-CD and AG-CD complex.
The thermograms of pure AG,γ
-CD,TPGS,physical mixture and AG-CD-TPGS ternary formulation were presented in Fig.4 A.AG presented a single endothermic peak at 239 °C,which indicated the melting point of crude AG.A broad endothermic peak at the range of 60–100 °C was shown forγ
-CD.This may be attributed to the dehydration process ofγ
-CD with the temperature increasing.TPGS exhibited a characteristic peak at 36.05 °C.The intensity of the AG melting peak weakened in the physical mixture and disappeared in the AG-CD-TPGS ternary formulation,which indicated much loss of its crystallinity and existed in the amorphous state.Fig.4 B showed the FTIR spectra of pure AG,γ
-CD,TPGS,physical mixture and AG-CD-TPGS ternary formulation at the wavelength of 400–4000 cm.The characteristic absorption peaks arising from the double bond of carbon (1675 cm),carbonyl (1727 cm) and methylene (2958 and 2979 cm)were observed at pure AG.However,the intensity and shape of these characteristic peaks weakened or disappeared in the AG-CD-TPGS ternary formulation.Furthermore,compared with the physical mixture,some typical peaks of AG in the AG-CD-TPGS ternary formulation also exhibited slight flattening,broadening and shifting.FTIR results indicated host CD molecules restricted the vibrating and bending of guest AG molecules due to the AG-CD complex formation.Also,both the lactone ring and decline ring in AG were likely inserted into the CD cavity based on the IR result,agreed with molecular modeling prediction.TheH NMR spectra of pure AG,γ
-CD and AG-CD-TPGS ternary formulation were shown in Fig.4 C.The presence of TPGS did not influence the spectra to the low concentration.The chemical shift of H-3 (3.942 ppm)and H-5 (3.862 ppm) inγ
-CD showed a significantly up-field shift to 3.909 ppm and 3.805 ppm,respectively.Because the proton of H-3 and H-5 was in the cavity of CD,when the guest molecule entered into the CD cavity,the chemical shift would have a noticeable change,which provided evidence of the formation of the complex [45] .Besides,compared with pure AG,the hydroxyl (5.152 ppm) in lactone ring and H-12 (7.025 ppm) of the AG-CD complex exhibited an upfield shift to 5.116 ppm and 6.899 ppm,respectively,which showed the lactone ring might enter the CD cavity.However,the H-19a (4.064 ppm) and H-19b (3.493 ppm) in the decalin ring also showed a slightly up-field shift to 4.046 ppm and 3.396 ppm.It seemed that the decalin ring might also reside in the CD cavity.In conclusion,the NMR results indicated the guest molecule (AG) inserted into the cavity of the host molecule (CD) and formed AG-CD complex.Both the lactone ring and the decalin ring had the opportunity to enter the cavity of CD and formed the mixture of two different types of complexes.The information on the structure of the AG-γ
-CD obtained by NMR validated the result of the MD simulation.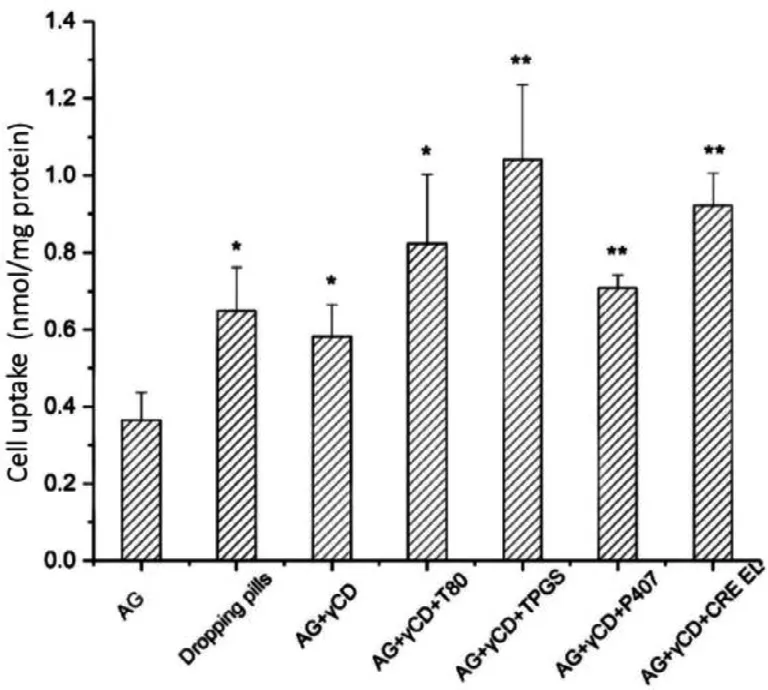
Fig.5–Influence of P-gp inhibitors on cellular uptake of AG by MDCK-MDR1 cells incubated at 37 °C for 90 min.∗P <0.05,∗∗P < 0.01 v.s.AG group.
The MD simulation was used as the in silico characterizing approach to explore the molecular mechanism of AGCDs before the physical characterization.It can provide the structural,dynamic and energetic information of the interaction between drugs and excipients.However,the MD simulation is limited by the accuracy of the force field,the starting model and computing capability.Thus,the MD simulation methods should be applied together with physical characterization analysis.
3.3.In vitro study
3.3.1.Cellular
uptake
P-gp excretion in the terminal intestine was another factor that limited the oral bioavailability of AG.Thus,the cellular uptake study aimed to screen the best-performing P-gp inhibitor among four representative inhibitors.The cellular uptake experiment results were shown in Fig.5 The results indicated that all the formulations and commercial dropping pills exhibited higher cellular uptake of AG compared with the AG group.In addition,the presence of four P-gp inhibitors increased the intracellular accumulation of AG.Among these four P-gp inhibitors,TPGS presented the most potency on increasing cellular uptake,which reached 1.04 nmol/mg protein.Therefore,TPGS was selected for the composition of the AG-CD-TPGS ternary system.
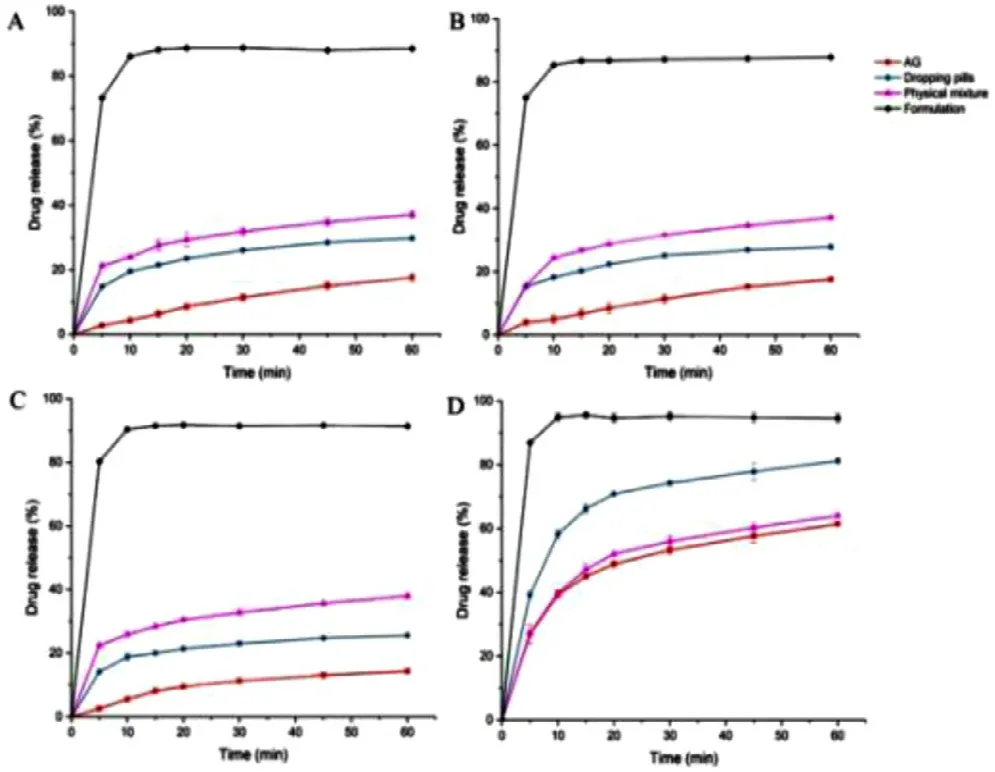
Fig.6–Dissolution profiles of pure AG,commercial dropping pills,physical mixture and AG-CD-TPGS ternary system in (A) 0.1 M HCl,(B) pH 6.8 PBS,(C) pure water and(D) water with 1% SDS.
3.3.2.Dissolution
study
Thein
vitro
drug release of pure AG,commercial dropping pills,physical mixture and AG-CD-TPGS ternary system in four different dissolution media were shown in Fig.6 .It was clear that the changing of pH did not exhibit a significant influence for AG release.Because of the low solubility of AG in distilled water,only 17% of pure AG released in different pH media.The accumulative releasing of the physical mixture was higher than pure AG,which may be attributed to the formation ofin situ
soluble complex in the dissolution medium.Commercial dropping pills enhanced AG release to nearly 40% to form solid dispersion and increased the specific surface area.AGCD-TPGS ternary system provides a significant improvement in dissolution rate,which showed the a 2.2-fold and 5.3-fold increase in drug release than commercial dropping pills and crude AG,respectively.All the samples showed higher drug release in the 1% SDS dissolution medium compared with other mediums.The SDS in the dissolution medium played a surfactant role and enhanced the dissolution rate.Briefly,the AG-CD-TPGS ternary system exhibited the highest drug release in all mediums,which contributed to inhibiting the crystallization of AG in dissolution progress.3.3.3.Transepithelial
transport
of
AG
To evaluate the effect of P-gp excretion,the transepithelial transport of crude AG,commercial dropping pills and AGCD-TPGS ternary system across the MDCK-MDR1 cells were administrated.The permeability coefficient and efflux ratio were summarized in Table 5 .The EfR value of 10.42 indicated a substantial efflux of AG in MDCK-MDR1 cell model at a concentration of 20 μM.Commercial dropping pills did not provide noticeable enhancement for the Pgp efflux.However,the secretory permeability coefficient(P app BL-AP 2.41 ±0.29 ×10cm/s) for the AG-CD-TPGS ternary system was only 4.15-fold than its absorptive permeability coefficient (P app AP-BL 0.58 ±0.04 ×10cm/s),which exhibited a significant inhibition for AG efflux.Furthermore,in comparing crude AG,the considerable difference between increasing absorption and decreasing efflux was obvious.It meant the new AG formulation had an effective influence on inhibiting the P-gp excretion and very likely enhanced the bioavailability for AG.
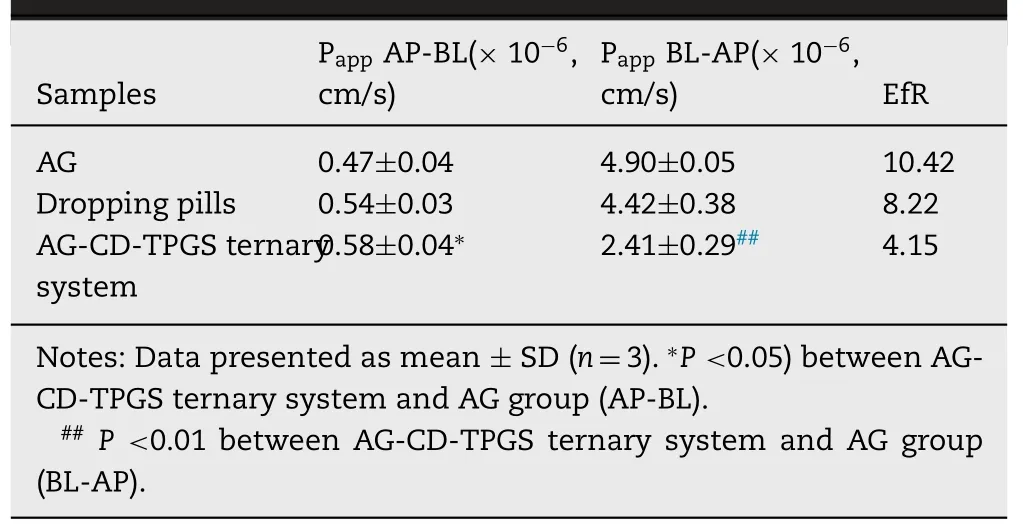
Table 5–Comparison of P app and EfR of AG (20 μM) across MDCK-MDR1 cell monolayer in AG,commercial dropping pills and AG-CD-TPGS ternary system.
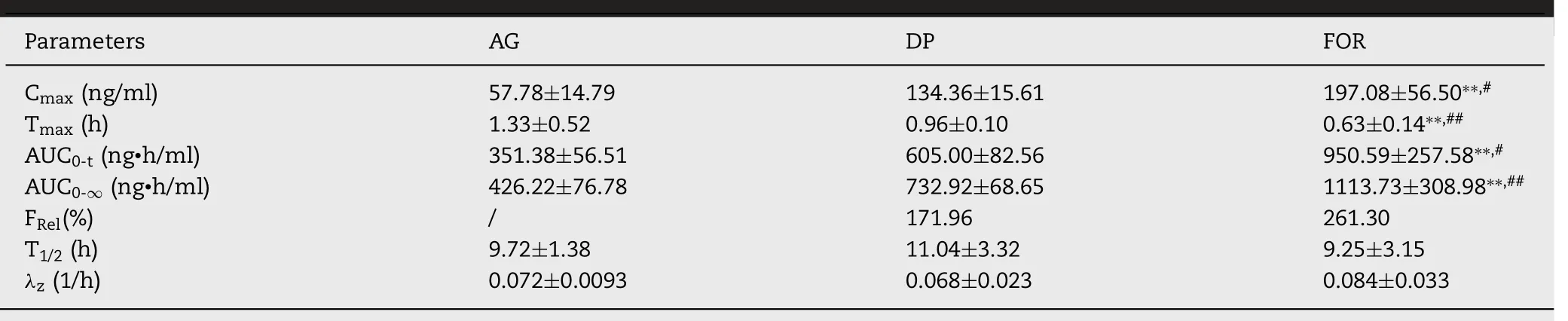
Table 6–Pharmacokinetic parameters of pure AG,commercial dropping pills and AG-CD-TPGS ternary system after oral administration of 40 mg/kg BW in Sprague-D
3.4.The physiologically based absorption modeling prediction
A desired pharmacokinetic behavior is expected in formulation design.PBAM can be used to predict the PK profile of candidate formulations.In the present work,the enhanced absorption is the supposedly dominant improvement of CD formulation.After absorption,the drug disposition,such as distribution,metabolism,and excretion,is out of the primary concern.Thus,a physiologically-based absorption model was used to handle the factors in formulations impacting the absorption,and the disposition was reduced to a threecompartmental model.This combined PBAM is commonly used in oral formulation investigations [3,46] .In the present work,the PBAM has been refined to consider the free drug fraction in CD formulation because only drug molecules that are not trapped in the CD’s cavity can be absorbed [28] .For the pure AG and dropping pill simulation,F
is always equal to 1 because no CD is administrated concomitantly,and absorption is based on fixed permeability.However,for the formulation containing CD,only drug molecules that are not trapped in the CD’s cavity can be absorbed [28] .Therefore,permeation is influenced by a drug-free fraction.In this model,F
is continuously calculated according to real-time CD concentration in each intestine segment during the simulation so that AG absorption,which can be judged as a product of permeability,andF
is adjusted continuously.Modeling for ternary formulation ignores the impact of TPGS because it was added to ameliorate the efflux effect of P-gp protein,but investigation [31] has found the P-gp does not show a significant effect on AG in the duodenum and the jejunum,which are the main site where AG is absorbed.The rat PBAM prediction of AG formulation using MATLAB SimBiology APP against observed data is shown in Fig.7 A–7H.After parameters fitted toi.v.
data,this model was used to simulate pure formulation.The initial result (not displayed)showed the predicted shape and peak of the curve was accurate,but the tail is higher than observed data,so colon transport rate constant was further fitted,and the result is given in Fig.7 A–7 H.After that,all parameters except for formulation type specific ones were maintained unchanged and simulate AG dropping pill and formulation with CD.Visual inspection shows this model has good accuracy,especially for CD formulation,wherein the simulated curve nearly goes exactly as the observed profile.Differences in many PK parameters (C
max,T
max andAUC
) are within a 1.2-fold range,indicating this predictive model is precise.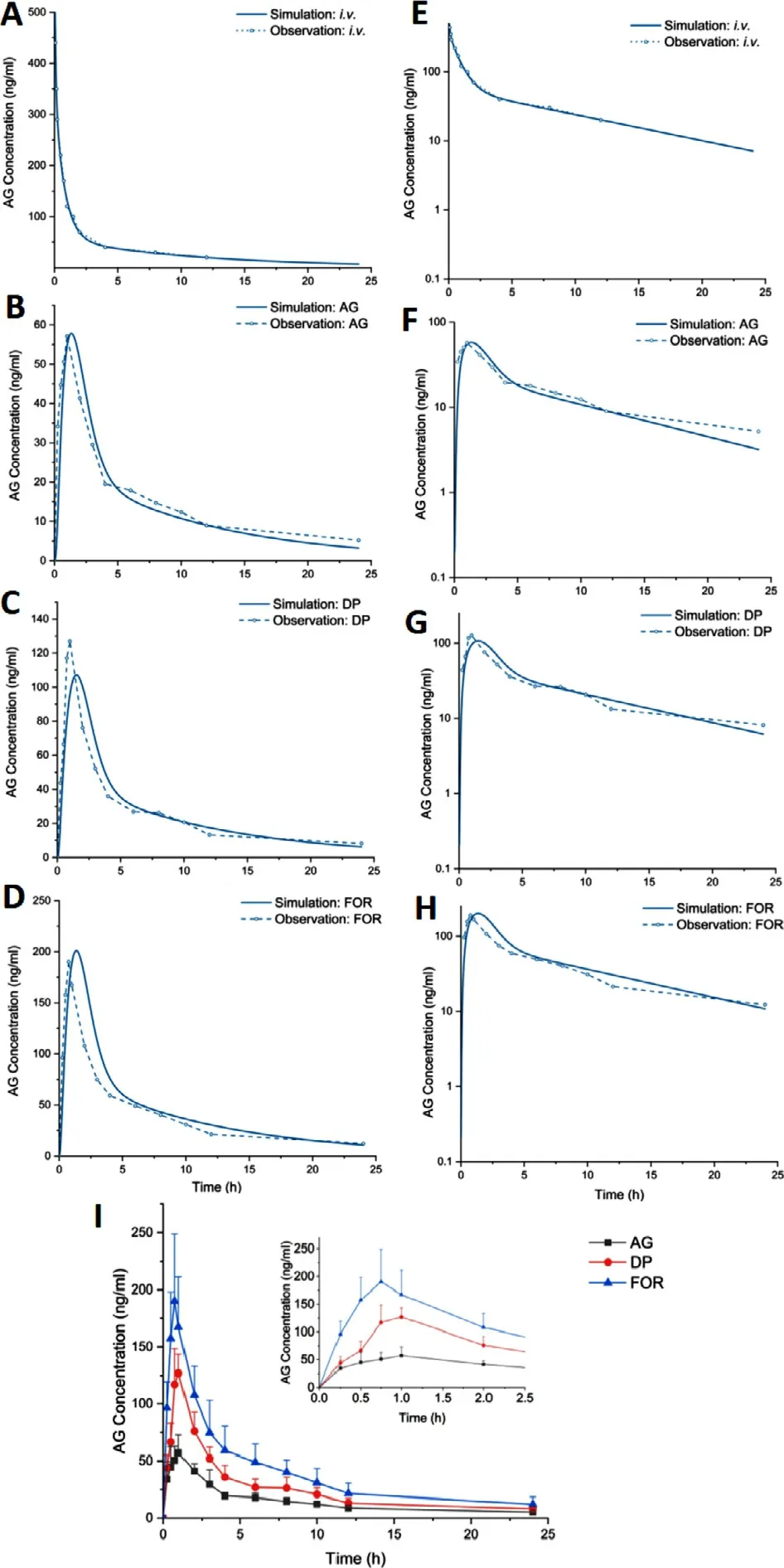
Fig.7–The comparison between observed PK points and simulated PK profiles along with fractional absorption of AG in some intestinal segments of (A) i.v.AG,(B) oral AG,(C)oral DP and (D) oral FOR in linear scale and (E) i.v.AG,(F)oral AG,(G) oral DP and (H) oral FOR in log scale;And (I)mean plasma concentration-time curve of pure AG,commercial dropping pills and AG-CD-TPGS ternary system after oral administration of 40 mg/kg BW in SD rats.Data presented as mean ±SD (n=6).(AG,andrographolide;DP,dropping pills;FOR,formulation).
3.5.In vivo pharmacokinetic study
Since the new AG formulation had increased the solubility and inhibited the P-gp excretion for AG,it was necessary to evaluate whether the AG-CD-TPGS ternary system could improve bioavailability after oral administration.The bioavailability of the AG-CD-TPGS ternary system was detected according to the pharmacokinetic study.The plasma drug concentration-time profiles of pure AG,commercial dropping pills and AG-CD-TPGS ternary system were shown in Fig.7 I,and the main pharmacokinetic parameters were summarized in Table 6 .The selected ion chromatogram for AG was shown in Fig.S2.C
of formulation (197.08 ng/ml)was significantly higher than those of pure AG (57.78 ng/ml)and commercial dropping pills (134.36 ng/ml).It might be attributed to the highest solubility of AG in the formulation,and more AG had been absorbed into the blood.Compared with pure AG and dropping pills,T
max of formulation exhibited an evident shortening due to the apparent enhancement of dissolution rate.Tand lambda z (λ
) value of the formulation were 9.25 h and 0.084 1/h.Also,the area under the curve from 0 h to infinity (AUC
0-
∞) of formulation (1113.73 ng • h/ml)also increased significantly with the comparison of pure AG(426.22 ng • h/ml) and dropping pills (732.92 ng • h/ml).Hence,the bioavailability of formulation was 2.60-fold and 1.59-fold than that of pure AG and dropping pills,respectively.4.Conclusion
In this study,the integrated computational tools had been applied to design drug-CDs complex formulation.The lightGBM prediction model for binding free energy between AG and CDs showed the same tendency with the experimental results.In traditional pharmaceutical formulation researches,characterization methods,such as DSC,FTIR and NMR,only can provide us static information about the state of drugs and excipients in formulations.As supplementary,the molecular modeling displayed the dynamic process between drug molecules and CD molecules at the atomistic level.According to the results ofin
vitro
experiments,the AGCD-TPGS ternary system could increase the solubility and dissolution rate significantly.Noticeable enhancement of cellular uptake of AG and decrease for P-gp efflux had also been demonstrated in the MDCK-MRD1 cell model.The PBAM of pure AG,AG dropping pills and AG-CD-TPGS ternary formulation showed consistent results within
vivo
pharmacokinetic study.This is the first time to integrate various computational tools to develop a novel AG-CD-TPGS ternary formulation with significant improvement of aqueous solubility,dissolution rate as well as the oral bioavailability.The integrated computational tool is a novel and powerful methodology to facilitate pharmaceutical formulation design.Conflict of interest
The authors report no conflicts of interest.
Acknowledgments
This work was financially supported by the FDCT Project 0029/2018/A1 and the University of Macau Research Grants(MYRG2019-00041-ICMS).This work was performed in part at the High-Performance Computing Cluster (HPCC) which is supported by Information and Communication Technology Office (ICTO) of the University of Macau.
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.ajps.2021.03.006 .
杂志排行

Asian Journal of Pharmacentical Sciences的其它文章
- An understanding of mitochondria and its role in targeting nanocarriers for diagnosis and treatment of cancer
- An insight into the in vivo imaging potential of curcumin analogues as fluorescence probes
- Recent advances in nanotherapeutics for the treatment and prevention of acute kidney injury
- Size,shape,charge and“stealthy”surface:Carrier properties affect the drug circulation time in vivo
- Macrophage membrane-mediated targeted drug delivery for treatment of spinal cord injury regardless of the macrophage polarization states
- The effect of ethanol on the habit and in vitro aerodynamic results of dry powder inhalation formulations containing ciprofloxacin hydrochloride