Stabilization of chromium(VI)by hydroxysulfate green rust in chromium(VI)-contaminated soils
2021-10-15LeilaALIDOKHTShahinOUSTANAlirezaKHATAEEMohammadNEYSHABURIandAdelREYHANITABARFacultyofAgricultureDepartmentofSoilScienceUniversityofTabrizTabriz5666647Iran
Leila ALIDOKHTShahin OUSTAN*Alireza KHATAEEMohammad R.NEYSHABURI and Adel REYHANITABARFaculty of AgricultureDepartment of Soil ScienceUniversity of TabrizTabriz 5666-647(Iran)
2Research Laboratory of Advanced Water and Wastewater Treatment Processes,Department of Applied Chemistry,Faculty of Chemistry,University of Tabriz,Tabriz 51666-16471(Iran)
3Department of Environmental Engineering,Gebze Technical University,Gebze 41400(Turkey)
ABSTRACT Chromium(Cr)-contaminated soils pose a great environmental risk,with high solubility and persistent leaching of Cr(VI).In this study,hydroxysulfate green rust(GRSO4),with the general formula Fe(II)4Fe(III)2(OH)12SO4·8H2O,was evaluated for its efficiency in Cr(VI)stabilization via Cr(VI)reduction to Cr(III)in four representative Cr(VI)-spiked soils.The initial concentrations of phosphate buffer-extractable Cr(VI)(Cr(VI)b)in soils 1,2,3,and 4 were 382.4,575.9,551.3,and 483.7 mg kg-1,respectively.Reduction of Cr(VI)to Cr(III)by structural Fe(II)(Fe(II)s)in GRSO4 in all studied soils was fast,wherein the application of GRSO4 markedly decreased the amount of Cr(VI)b at the Cr(VI)b/Fe(II)s stoichiometric mole ratio of 0.33.The kinetics of Cr(VI)reduction by GRSO4 could not be determined as this reaction coincided with the release of Cr(VI)from soil during the experiment.The concentration of Cr(VI)b decreased,as the Cr(VI)b/Fe(II)s ratio decreased from 0.46 to 0.20,generally to below 10 mg kg-1.Back-transformation of the generated Cr(III)was examined in the presence of manganese oxide birnessite at the birnessite/initial Cr(III)mole ratio of 4.5.The results of batch tests showed that only 5.2%of the initial Cr(III)was converted to Cr(VI)after two months,while under field capacity moisture conditions,less than 0.05%of the initial Cr(III)was oxidized to Cr(VI)after six months.The results illustrated that remediation of Cr(VI)-contaminated soils would be fast,successful,and irreversible with an appropriate quantity of fresh GRSO4.
Key Words:batch system,birnessite,Cr(III)oxidation,layered double hydroxide,reduction,soil remediation
INTRODUCTION
Chromium(Cr)contamination in soil refers to the accumulation of Cr due to anthropogenic activities,which greatly affects the main chemical and microbial functions of the soil and,subsequently,impacts the quality of groundwater.In Iran,Cr contamination arises mainly from active chromite(FeCr2O4)mining sites,which are located in the northeastern region of Iran,and the Cr plating industry(Jafarian and Jafarian,2017).Chromium exist in several oxidation states(-II to+VI),but only hexavalent(Cr(VI))and trivalent(Cr(III))forms are thermodynamically stable in natural systems(Kotaśand Stasicka,2000).Oxidation,reduction,sorption,hydrolysis,and precipitation of these two Cr oxidation states in soils are markedly influenced by the soil redox potential,pH,organic carbon(OC)content,metal levels,cation exchange capacity(CEC),and competing ions.In the soil environment,Cr(VI)can be reduced to Cr(III),which is involved in reactions with organic matter,ferrous ion(Fe(II)),and bisulfide(HS-)ion.Oxidation of Cr(III)to Cr(VI)occurs naturally in soils high in manganese(Mn(IV))oxides(Shahidet al.,2017).
Inherently,Cr(VI)is highly toxic,carcinogenic,and mobile in soils.Chromium(VI)species(e.g.,HCrO-4,CrO2-4,and Cr2O2-7)are not appreciably retained by soils due to the repulsive forces of negatively charged clay minerals.They are weakly sorbed on metal oxide surfaces in soils at acidic conditions or are reduced to Cr(III)by soil organic matter(OM)and Fe(II)-bearing minerals(Hsuet al.,2015;Veselskáet al.,2016;Zhanget al.,2019).In contrast,Cr(III)is nontoxic and virtually immobile due to its strong adsorption on soil particles and tendency to precipitate as Cr(OH)3or co-precipitate as the solid solution Cr(III)/Fe(III)/OH in the presence of Fe(III)(Norseth,1981).
Hence,chemical reduction of Cr(VI)to Cr(III)is an acceptable and effective technique for decreasing the toxic effects of Cr(VI)and its stabilization in polluted sites.Several reductants have been proposed for remediation of Cr(VI)-contaminated soils and waters,including OC(Sunet al.,

2009;Aldmouret al.,2019),sodium thiosulfate(Kostareloset al.,2009),zerovalent iron nanoparticles(Reyhanitabaret al.,2012;Gaoet al.,2018),iron scrap(Hoseiniet al.,2015),and Fe(II)iron(Tinjumet al.,2008;Liuet al.,2018;Wanget al.,2019).Among the various reductants used,Fe(II)iron,in the form of aqueous Fe(II)(Fe(II)aq)or solid-bound Fe(II)in minerals,is the strongest and fastest reductant of Cr(VI).Reduction of Cr(VI)to Cr(III)by Fe(II)aqcan be expressed as the following overall reaction(Eary and Rai,1988):Green rust(GR),belonging to the layered double hydroxide(LDH)family,consists of positively charged brucite-like mixed Fe(II)/Fe(III)hydroxide layers and planaror tetrahedralanions and water molecules in the interlayer space.The general formula of GR is(Fe(II)1-xFe(III)x(OH)2)x+·(x/nAn-·m/nH2O)x-,where x is the molar fraction of Fe(III),An-donates the intercalated anion,and m is a varying amount of interlayer water(Génin and Ruby,2004).This compound was initially recognized as an intermediate species in the corrosion of iron.Green rust has also been naturally identified in hydromorphic soil and Fe(II)-rich groundwater(Schwertmann and Fechter,1994).The large Fe(II)fraction in the structure of GRs makes them strong reductants,which may contribute to the removal of redox-sensitive compounds such as chlorinated hydrocarbons(Lee and Batchelor,2002),heavy metals(Bond and Fendorf,2003),metalloids(Rahmanet al.,2012),and nonmetals(Hansenet al.,2001).The efficiency of structural Fe(II)(Fe(II)s)in LDHs for the removal of Cr(VI)from aqueous solutions has been studied by several researchers(Bond and Fendorf,2003;Alidokhtet al.,2016,2018;Heet al.,2017).The hydroxysulfate GR(GRSO4)reduces Cr(VI)to Cr(III)according to the following reaction,and produces relatively insoluble Cr(III)/Fe(III)oxyhydroxide(Skovbjerget al.,2006):
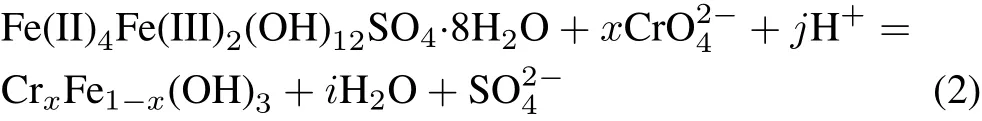
Green rust offers a number of advantages over Fe(II)aqas a reductant of Cr(VI).i)In contrast to dissolved Fe(II),which generates net acidity during the reduction of Cr(VI)to Cr(III)(Eq.1),the reduction of Cr(VI)by GR results in an increased pH and,therefore,the availability of Cr(III)for oxidation decreases(the stability of the end product increases significantly by increasing the pH of the system(Palmer and Wittbrodt,1991)).ii)According to Pourbaix diagrams of iron species,GR is more stable than Fe(II)aqat higher pH values and a constant redox potential(Eh).Ferrous iron rapidly oxidizes and precipitates in alkaline environments,and becomes unavailable for the reduction of Cr(VI)(Geelhoedet al.,2003).iii)The Eh values calculated for half-cells comprising Fe(II)-Fe(III)reveal that GR is a stronger reductant than Fe(II)aq(Eqs.3 and 4)(Wagmanet al.,1982;Géninet al.,1998;Skovbjerget al.,2006).
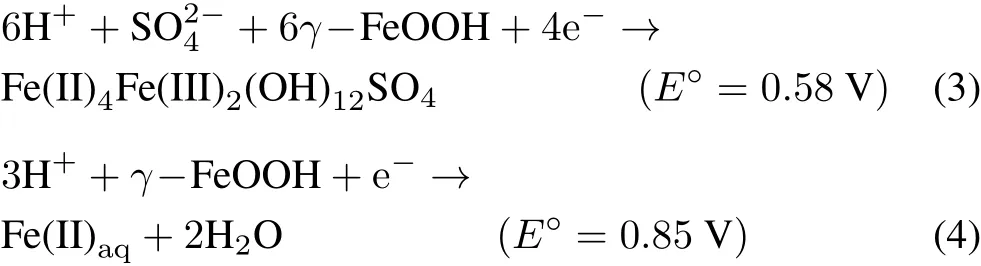
where E°is the standard electrode potential.however,the fate of generated Cr(III)is of environmental concern as it poses a potential danger if oxidized to Cr(VI).Manganese oxides are the only naturally occurring oxidizing agent of Cr(III)in soils(Bartlett and James,1979),and oxidize Cr(III)to Cr(VI)viathe following reaction(Fenget al.,2007):
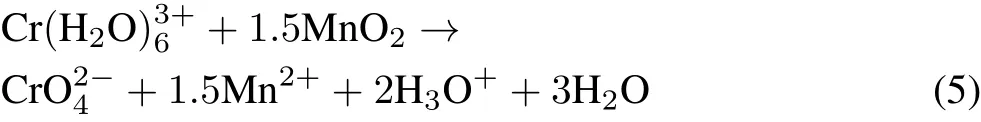
The aims of the present work were as follows:as the sorption and redox reactions of Cr in soils are generally controlled by their physico-chemical properties(Kožuhet al.,2000),our first goal was to evaluate the natural capacity of four selected soils for the stabilization of added Cr(VI).Next,we investigated the efficiency of GRSO4for reductive stabilization of Cr(VI)in Cr(VI)-spiked soils under various experimental conditions.Finally,to ensure the irreversibility of the reaction,we studied the possibility of back-transforming the generated Cr(III)to Cr(VI)in the presence of manganese oxide birnessite(δ-MnO2)under batch and field capacity(FC)moisture conditions.
MATERIALS AND METHODS
Synthesis of GRSO4 and δ-MnO2
According to a co-precipitation method(Refaitet al.,1998;Rubyet al.,2003),GRSO4was synthesized in a 1-L four-neck glass reactor under nitrogen gas flow.Deoxygenated distilled water was used to prepare the solutions.The synthesis method involved the slow addition(100 μL min-1)of NaOH solution to a mixed solution of Fe(II)(as FeSO4·7H2O)and Fe(III)(as Fe2(SO4)3·6H2O)at room temperature.The total concentration of Fe(II)+Fe(III)in the solution was 0.05 mol L-1(confirmed by atomic absorption spectroscopy(AAS)analysis).Solution of NaOH was added to produce an OH/Fe ratio of 2 and to achieve a required final concentration of 0.1 mol L-1NaOH.During the addition of NaOH solution,the suspension was vigorously stirred with a magnetic stirrer.The mole fraction of Fe(III)was set to 0.33.The final pH of the resultant suspension was 7.5±0.2.
In the present study,δ-MnO2was used as an oxidant of Cr(III).Birnessite was synthesized according to the method of Fenget al.(2007).Briefly,500 mL of 0.4 mol L-1KMnO4solution was heated in an oil bath and maintained at boiling with stirring.A mixed solution of concentrated HCl(35 mL)and distilled water(15 mL)was added to the boiling KMnO4solution dropwise.The solution was stirred for a further 30 min and then aged for 12 h.Precipitates were separated by centrifugation at 9 300×g for 5 min,washed with distilled water,and dried at 70°C.All chemicals were of analytical grade with a purity of 97%or greater.
Samples were characterized by X-ray diffraction(XRD),using a Bruker D5000 powder X-ray diffractometer(Siemens,Germany)with CuKαradiation at 40 kV and emission current of 30 mA.Freshly prepared GR was separated from its solution by centrifugation at 9 300×g for 2 min.To inhibit oxidation of the sample during analysis,the precipitates were mixed with glycerol(99.5%)under nitrogen atmosphere and the prepared paste was smeared onto a glass slide for XRD analysis at 4°–70°2θ.Peak baseline was subtracted using OriginPro software.Scanning electron microscopy(SEM)images were obtained using a Mira3-Tescan(TESCAN Co.,Czech Republic)at an acceleration voltage of 20 kV to examine the size and morphology of the synthesized GRSO4.For SEM analysis,the GR was vacuum-dried after filtration and loaded onto a carbon(C)stub under nitrogen atmosphere.Then,the sample was coated with gold in a vacuum deposition chamber before analysis.In addition,the point of zero charge(PZC)of the synthesized birnessite was determined through the potentiometric titration method(Appelet al.,2003).
To determine the Fe(II)concentration in the GRSO4structure,the unfiltered sample(GRSO4+aqueous phase)and the solid phase directly after filtration were dissolved in HCl solution(pH≈3).The concentration of Fe(II)in the unfiltered sample was used to estimate the amount of Fe(II)in aqueous phase by a mass balance approach.The Fe(II)concentration was measured colorimetrically using the 1,10-phenanthroline method(Schilt,1969).Sulfate was determined using the turbidimetry method(Sheenet al.,1935).
In the present work,a freshly prepared GR suspension was used without washing and drying to avoid the possible oxidation of GR.The density of GR in the suspension was determined by measuring the volume of suspension aliquots and weighing the solids after drying under vacuum conditions(Skovbjerget al.,2006).
Soil samples
Four uncontaminated soils with desired variation in the relevant properties were collected to a depth of about 30 cm from Paresar(soil 1),Fooman(soil 2),Tabriz(soil 3),and Espiran(soil 4)areas located in the north and northwest of Iran.Samples were air-dried and gently ground to pass through a 2-mm stainless steel sieve.The pH of the soil samples was measured in 1:1 soil:distilled water suspensions(McLean,1982);electrical conductivity(EC)was determined in 1:1 soil:distilled water extracts(Smith and Doran,1996);OC content of soils was measured by the Walkley-Black method(Nelson and Sommers,1996);soil texture was determined by the hydrometer method(Gee and Or,2002);calcium carbonate equivalent(CCE)percentage was determined by the acid-base titration method(Jackson,1958);cation exchange capacity was measured by the Bower method(Boweret al.,1952);FC moisture content was measured by the pressure plate method at 30 kPa(United States Salinity Laboratory Staff,1954).The adsorbed Cr(VI)in soil samples was extracted by phosphate buffer(K2HPO4/KH2PO4,1.2 mol L-1,pH 7.2)(James and Bartlett,1983)and the concentration of Cr(VI)was determined by the diphenylcarbazide colorimetric method with a detection limit ofca.0.1 μmol L-1(USEPA,1992)at 540 nm using a UV/Vis spectrophotometer(UV-9200,BFRL Co.,China).The total Cr(VI)and Cr(VI)+Cr(III)in the samples were extracted by alkaline digestion and aqua-regia procedures(Nieuwenhuizeet al.,1991;USEPA,1996),respectively.The Cr concentration in the soil extract was then measured using AAS(AA 3600,Shimadzu Co.,Japan).The concentration of easily reducible manganese in soil was determined according to the Gambrell and Patrick method(Gambrell and Patrick,1982).
Natural capability of soils for stabilization of Cr(VI)
In preliminary experiments,soils showed different Cr(VI)stabilization capabilities;therefore,each soil was spiked with different concentrations of Cr(VI).In a plastic container,500 g soil was sprayed with K2CrO4solution while mixing to reach levels of 1 000,2 500,4 000,and 5 500 mg Cr kg-1in soil 1,400,700,1 000,and 1 300 mg Cr kg-1in soil 2,and 400,600,800,and 1 000 mg Cr kg-1in soils 3 and 4.Then,soil moisture was adjusted to the FC conditions by adding distilled water.The soil samples were subjected to six wetting(FC moisture)-drying(air-dry)cycles at 26±0.5°C to simulate field conditions.Each cycle lasted for one week.To determine the equilibrium time of Cr(VI)stabilization,for the treatment with the highest Cr(VI)level of each soil,Cr(VI)was extracted with phosphate buffer solution at the end of each cycle.To evaluate the capacity of each soil for Cr(VI)stabilization,the concentration of buffer-extractable Cr(VI)was determined at equilibrium time at different initial concentrations of Cr(VI).The Cr(VI)stabilization efficiency of each soil(SEsoil,%)was calculated as follows:
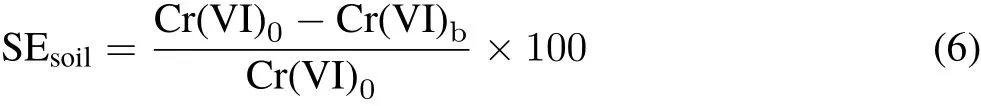
where Cr(VI)0is the initial concentration of Cr(VI)(mg kg-1)added to the soils and Cr(VI)bis the phosphate buffer-extractable Cr(VI)(mg kg-1)of the soils after four wetting-drying cycles.
Preparation of Cr(VI)-spiked soils for batch experiment
We tried to evaluate the efficiency of Cr(VI)stabilization process in a relatively constant initial concentration of Cr(VI).For this purpose,the soil samples were spiked with K2CrO4solution to attain a Cr concentration(in the form of Cr(VI)b)around the chosen value of 450 mg kg-1.The initial concentration of Cr(VI)was calculated from the linear regression curve fitted to the data of initial and final concentrations of Cr(VI)(Mahdiehet al.,2016)obtained from previous section.A proper volume of K2CrO4solution with a desired concentration was then added to 500 g of air-dried soil.The soils were moistened with a necessary volume of distilled water to reach the FC moisture condition.The Cr(VI)-spiked soils were mixed thoroughly and subjected to four wetting-drying cycles at room temperature.The soil samples were then air-dried,homogenized,and sieved(<2 mm).The concentration of Cr(VI)b,total Cr(VI),total Cr(Cr(III)+Cr(VI)),pH,and EC were determined for the Cr(VI)-spiked soils.
Batch experiment of Cr(VI)stabilization by GRSO4
The experiment was conducted using 50-mL centrifuge tubes with 0.01 mol L-1K2SO4as the background solution and without pH adjustment.During the experiment,the presence of oxygen was not prevented.The experiment was carried out in duplicate,and mean values are presented.
Effects of soil pretreatment and reaction time on Cr(VI)stabilization efficiency
These experiments were performed with pretreated and non-pretreated soils at Cr(VI)b/Fe(II)smole ratio of 0.33,where Fe(II)sis the amount of structural Fe(II)in GRSO4.The soil/solution ratio was 0.1;that is,the total volume of background solution plus GRSO4suspension was 20 mL and the soil mass(on oven-dried basis)was 2 g.
The experiment with pretreated soil samples(soaked samples)was carried out as follow:2 g of dry soil was weighed into a centrifuge tube and a certain volume of the background solution(0.01 mol L-1K2SO4)was added.The suspensions were kept in an incubator at 26±0.5°C for 24 h.Then,GRSO4suspension was added to the soil suspension and shaken on a reciprocal shaker at 120 r min-1.The volume of the GRSO4suspension was determined according to the required concentration of Fe(II)sin each experiment.At various time intervals,2 mL aliquots were withdrawn and immediately filtered through a 0.22-μm syringe filter.Then,the concentration of Cr(VI)in the filtered solution was determined.In the experiment with non-pretreated soil samples(air-dried samples),the background solution(0.01 mol L-1K2SO4)was added simultaneously with the GRSO4suspension to the soils.
To estimate the concentration of Cr(VI)that may be available for GRSO4during the reaction,experiments were conducted without GRSO4to account for Cr(VI)released from soil in the presence of K2SO4solution(0.01 mol L-1).For this,20 mL of K2SO4solution was added to 2 g of the Cr(VI)-spiked soils,which were then shaken at 120 r min-1.The concentration of Cr(VI)in the filtered solution was determined at different time intervals.
Another set of experiments were conducted to determine the concentration of Cr(VI)bat the end of the reaction between Cr(VI)in the Cr(VI)-contaminated soils and GRSO4.For this,soil samples were reacted with GRSO4for 120 min and then 2 mL of the buffer solution was added to the suspension and shaken for a further 120 min.The suspension was then centrifuged at 4 100×gfor 5 min and filtered through a 0.22-μm syringe filter.Then,the concentration of Cr(VI)in the filtered solution was determined.The Cr(VI)stabilization efficiency by GRSO4(SEGR,%)was calculated using Eq.7:

where Cr(VI)Rbis the phosphate buffer-extractable Cr(VI)(mg kg-1)after the soil reacted with GRSO4.
Effect of Cr(VI)b/Fe(II)s mole ratio on Cr(VI)stabilization efficiency
The soaked soil samples were reacted with GRSO4at Cr(VI)b/Fe(II)sratios of 0.20,0.33,and 0.46 for 60 min at a soil/solution ratio of 0.1.Then,the suspensions were centrifuged at 4 100×gfor 5 min and the supernatant was analyzed for dissolved Cr(III)and final pH.In separate batches,Cr(VI)Rbwas measured in GRSO4-treated soil samples.
Effect of soil/solution ratio on Cr(VI)stabilization efficiency
To examine the effect of soil/solution ratio,experiments were carried out in soil 3,at Cr(VI)b/Fe(II)sratio of 0.33 and soil/solution ratios of 0.5,0.2,0.1,0.05,and 0.02.The total volume of background solution plus GRSO4suspension was 20 mL,and soil weights were 10,4,2,1,and 0.4 g,respectively.The soaked soil samples were reacted with GRSO4for 60 min,and Cr(VI)Rbwas determined.
Back-transformation of produced Cr(III)
Back-transformation of produced Cr(III)to Cr(VI)in the presence of birnessite was assessed in soil 4.In a set of batch experiments,17.2 mL of K2SO4solution(0.01 mol L-1)was added to a centrifuge tube containing 2 g soil and 7.4 mg birnessite.The suspension was equilibrated at 26±0.5°C for 24 h.Then,2.8 mL of freshly synthesized GRSO4suspension was added to the tubes to produce a Cr(VI)b/Fe(II)sratio of 0.2.The mixture was shaken for 60 min at 120 r min-1and then transferred into an incubator at 26±0.5°C.The suspensions were periodically shaken(60 min a day)and the concentration of the produced Cr(VI)was determined by the phosphate buffer procedure at different intervals(1–60 d).The maximum concentration of Cr(III)that could react with birnessite was 18.92 μmol L-1for 2 g of soil(17.4 μmol L-1produced from the reduction of Cr(VI)by GRSO4and 1.52 μmol L-1from the reduction of Cr(VI)by soil during wetting-drying cycles).To provide excess amounts of oxidant in the reaction system,birnessite was added to the soil at a molar ratio ofδ-MnO2/Cr(III)=4.5.
The experiments were repeated in FC moisture to simulate the field conditions.One hundred grams of the soil sample were homogeneously mixed with 0.37 mg of birnessite and FC moisture was reached with the addition of K2SO4solution(0.01 mol L-1).The mixture was stored at 26±0.5°C for 24 h.Then,48 mL of fresh GRSO4suspension was added.In these experiments,GRSO4was prepared at a total Fe concentration of 0.15 mol L-1to decrease the volume of required GR suspension.The soil samples were stored in an incubator at 26±0.5°C and the moisture level was allowed to decrease and then maintained constant at FC conditions by periodical weighing and watering.At different time intervals(1 d to 6 months),FC moist soil samples(equivalent to 2.0 g of oven-dried soils)were weighed into 50 mL centrifuge tubes,and 20 mL of 0.01 mol L-1K2SO4solution and 2 mL of phosphate buffer solution were added.Then,the suspensions were shaken for 120 min and centrifuged at 4 100×gfor 5 min,and the concentration of Cr(VI)was measured.All experiments were carried out in duplicate,and the reported results are the mean values.
Analysis of the reaction final product in a pure system
X-ray photoelectron spectroscopy(XPS)and XRD techniques were used to analyze the product of the reaction between Cr(VI)and GRSO4in an aqueous solution as a pure system;Cr(VI)reacted with GRSO4at a stoichiometric ratio of 0.33(Cr(VI)/Fe(II)s)until equilibrium was reached.Reddish-brown precipitates were separated from solutionviacentrifugation and vacuum-dried.The filtrate was analyzed for dissolved Cr(III)and Fe(II).The Fe(II)content was also determined in the solid precipitates.Then,precipitates were analyzed by XPS(EA10 Plus,Bestec Co.,Germany)with a rotating aluminum anode as the X-ray source,and XRD analysis of the final product was performed in a 2θrange of 7°–70°.
RESULTS AND DISCUSSION
Characterization of GRSO4 and δ-MnO2
The concentration of the synthesized GRSO4andits constituent elements are presented in Table I.The Fe(III)/(Fe(II)+Fe(III))and Fe(II)/Fe(III)mole ratios in the GRSO4structure were 0.32 and 2.12,respectively,which correspond to the values defined for GRSO4(Rubyet al.,2003,2010).
The XRD pattern of GRSO4is shown in Fig.1.The observed diffraction peaks at 7.92°,16.02°,24.16°,32.29°,and 33.30°are characteristic of GRSO4.No impurities in the samples were detected by XRD.The average crystal size of GRSO4calculated from the XRD data and using Scherrer equation(Cullity,1978)was 18.15 nm.Additionally,the interlayer space(d-spacing)of the GRSO4crystal,obtained from the XRD data,was 1.115 nm,which is identical to that reported for GRSO4(Guilbaudet al.,2013).The SEM images of the freshly prepared GRSO4are represented in Fig.2,exhibiting hexagonal morphology of the platelets.
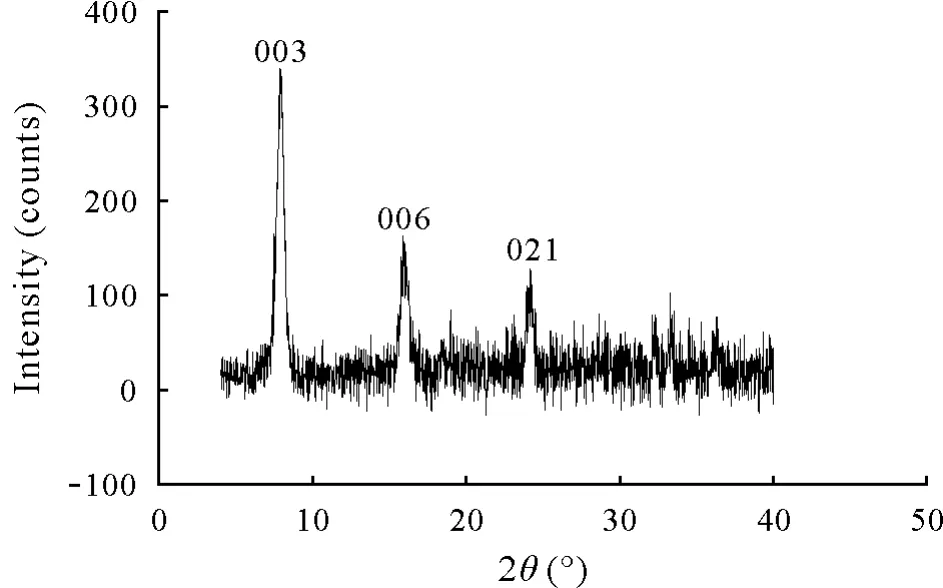
Fig.1 X-ray diffraction(XRD)pattern of the freshly prepared hydroxysulfate green rust(GRSO4).Values above the peaks indicate the corresponding Miller indices in the hexagonal structure.

TABLE IConcentration and chemical compositiona)of the synthesized hydroxysulfate green rust(GRSO4)

Fig.2 Scanning electron microscopy(SEM)images of the freshly prepared hydroxysulfate green rust(GRSO4).
The XRD pattern of the synthesized birnessite(Fig.S1,see Supplementary Material for Fig.S1)showed the typical diffraction pattern of birnessite(Fenget al.,2007).The pHPZCof birnessite was 4.2,indicating that its surface would be positively charged at pH values below this otherwise negatively charged.
Soil characteristics
Selected properties of the uncontaminated soil samples used in this study are presented in Table II.It can be seen that soil 1 is an acid soil with high OC content;soil 2 is an acid soil with low OC content;soil 3 is an alkaline,slightly calcareous soil(CCE=4.5%)with low OC content;while soil 4 is an alkaline,calcareous soil(CCE=18%)with low OC content.
Natural capability of the soils to stabilize Cr(VI)
Knowledge on the equilibration time for Cr(VI)reduction by soil was necessary to ensure that the concentration of Cr(VI)is not changed due to its reaction with soil OC during the batch removal experiments.Changes in the Cr(VI)bconcentration of the studied soil samples during the six wetting-drying cycles are shown in Fig.3.The results showed that an equilibrium time beyond four cycles(one month)was obtained for all four soils.The initial and final concentrations of Cr(VI)in the soils,as well as the Cr(VI)SEsoilvalues,are presented in Table III.

Fig.3 Phosphate buffer-extractable Cr(VI)(Cr(VI)b)from the four soils during six wetting-drying cycles.Bars are standard errors(n=3).
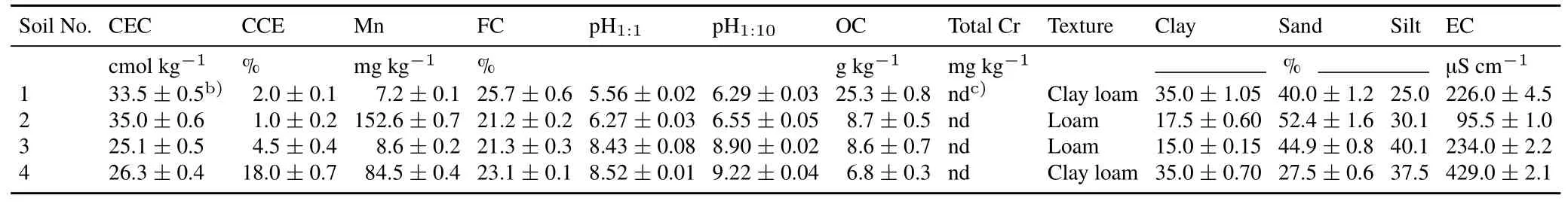
TABLE IISelected physico-chemical propertiesa)of the uncontaminated soil samples used in this study

TABLE IIIInitial and final concentrations of Cr(VI)(Cr(VI)0 and Cr(VI)r,respectively)as well as the Cr(VI)stabilization efficiencies(SEsoil)of the four soils after four wetting-drying cycles
The results indicated that the ability of the studied soils to remove Cr(VI)was in the following order:soil 1>soil 2≈soil 3>soil 4.Soil samples with high OC content and low pH value had high Cr(VI)stabilization efficiencies.This is because low soil pH and high OC content enhance the stabilization of Cr(VI)in soil by driving its reduction to Cr(III)through the following reaction(Kožuhet al.,2000):
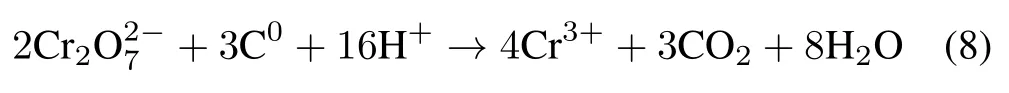
Our results are consistent with the findings of other researchers(Jardineet al.,2013),who investigated the influence of soil geochemical and physical properties on the sorption and bioaccessibility of Cr(VI)in 35 soil samples.Those researchers found that Cr(VI)sorption was high in soils with a high total organic carbon(TOC)content and a low pH.In addition,the bioaccessibility of Cr(VI)in studied soils decreased with increasing soil TOC content.
The concentration of immobilized Cr(VI)increased,while the Cr(VI)stabilization efficiency decreased significantly as the initial concentration of Cr(VI)increased.An increase in the initial concentration of Cr(VI)has been shown to considerably enhance the rate of Cr(VI)reduction by soil humic substances.The enhanced reduction rates resulted in the increasing immobilized concentration of Cr(VI)(Wittbrodt and Palmer,1997).
Stabilization of Cr(VI)by GRSO4
The properties of the Cr(VI)-spiked soil samples prepared for batch stabilization experiments are presented in Table IV.The quantities of Cr(VI)extracted by distilled water(dissolved form)comprised the largest percentage of the buffer-extractable Cr(VI)(sum of dissolved and exchangeable forms)and the total Cr(VI)(all extractable forms)in alkaline soils.The lower soil pH could create more positive charge on the soil OM and mineral surfaces(e.g.,aluminum and iron oxides)(Jianget al.,2008),resulting in a stronger retention of Cr(VI)by the soil and a subsequent reduction in the concentration of water-extractable Cr(VI)in acid soils.
Effects of soil pretreatment and reaction time on Cr(VI)stabilization efficiency
Changes in Cr(VI)concentration in soil solution during reaction with GRSO4,and time-dependent release of Cr(VI)in the presence of K2SO4solution(0.01 mol L-1)are presented in Figs.4 and 5,respectively.In all studied soil samples,an initial rapid decrease in the concentration of aqueous Cr(VI)was observed after the addition of GRSO4solution,which was followed by a gradual increase.During the stabilization experiments,two reactions occurred at different rates.Reaction 1 was the reduction of Cr(VI)by GR,which occurred at a high rate and minimized the Cr(VI)concentration in soil solution during the first few minutes.Reaction 2 involved the release of Cr(VI)from soil particles,which proceeded slower than reaction 1.Although,these experiments were performed at a stoichiometric ratio of Cr(VI)b/Fe(II)s=0.33,GR was oxidized before the complete release of Cr(VI)from soil,and its action was terminated.Therefore,the concentration of Cr(VI)in soil solution increased due to its gradual release from soil.Based on these results,we can conclude that the reduction of Cr(VI)by GRSO4is a rapid process,while at least one week is required for the efficient immobilization of Cr(VI)in contaminated soil when using FeSO4,FeS particles(Dermataset al.,2006;Moonet al.,2009;Liet al.,2017),and CaSx(Chrysochoouet al.,2010).
Liet al.(2017)investigated the efficiency of stabilized FeS particles and FeSO4in the reductive immobilization of Cr(VI)in a Cr-spiked soil.The molar ratio of Cr(VI)to FeS/FeSO4was 0.66 and the moisture content of soil was 50%.They reported that approximately 98% and 34% of Cr(VI)in soil was immobilized after 7-d incubation in the presence of stabilized FeS particles and FeSO4,respectively.In addition,the soil moisture content varied from 40%to 70%and the amount of natural OM in soil,when added to the Cr-spiked soil as humic acid at 1%–5%,had no significant effect on Cr(VI)immobilization by stabilized FeS particles.
In the present study,it was not possible to investigate the reduction reaction kinetics because of interference of Cr(VI)release from soil.In both pretreated and non-pretreated conditions,SEGRin the soils was in the following order:soil 4>soil 3>soil 2>soil 1.The percentages of stabilized Cr(VI)under pretreated conditions were 94.3%,94.7%,98.2%,and 99.7%in soil 1,soil 2,soil 3,and soil 4,respectively.Values under non-pretreated conditions were 89%,90.6%,97.1%,and 99.3%,respectively.As shown in Fig.5,the rate of chromate release from acid soil samples was lower than that from alkaline soil samples.Accordingly,less Cr(VI)was available in soil solution when GR was reactive,which lowered the stabilization efficiency in acid soils.Indeed,soil pH influences Cr(VI)stabilization efficiency by influencing Cr(VI)availability in soil solution.The stabilization effi-ciency of Cr(VI)in the pretreated conditions was higher than that in the non-pretreated conditions due to the high availability of Cr(VI)in soil solution.

Fig.5 Changes in Cr(VI)concentration with time in the four soils during their reactions with K2SO4 solution(0.01 mol L-1)at soil/solution ratio of 0.1.The four soils were pretreated(i.e.,soaked with 0.01 mol L-1 K2SO4)or not before the reaction.Bars are standard errors(n=3).

TABLE IVConcentrations of various forms of Cr,pH,and electrical conductivity(EC)of the Cr(VI)-spiked soils used for the batch stabilization experiment
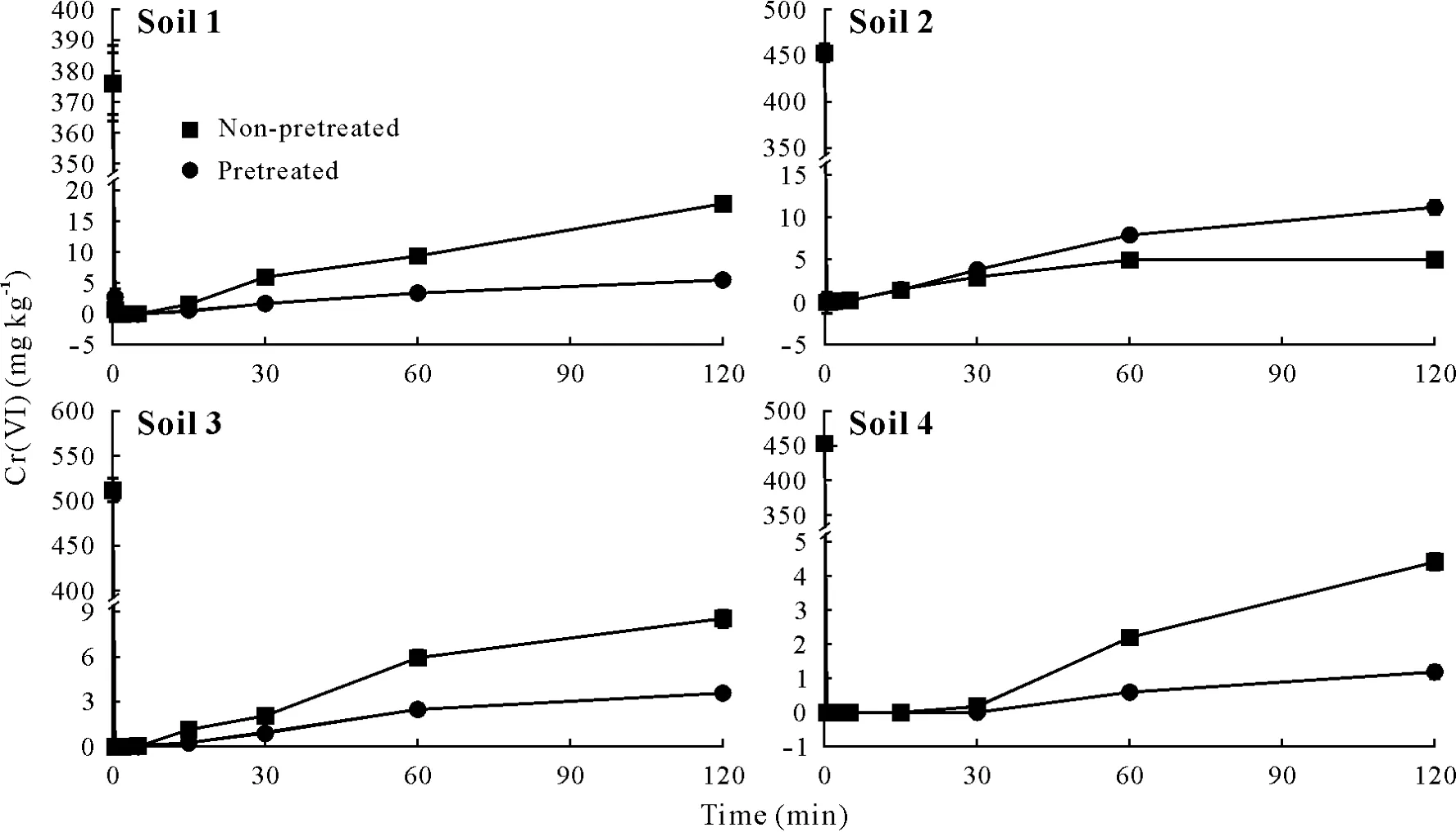
Fig.4 Changes in Cr(VI)concentration with time in soil solution during the reactions between the four soils and hydroxysulfate green rust(GRSO4)at soil/solution ratio of 0.1 and Cr(VI)b/Fe(II)s mole ratio of 0.33,where Cr(VI)b is the phosphate buffer-extractable Cr(VI)of the soils after four wetting-drying cycles and Fe(II)s is the amount of structural Fe(II)in the GRSO4.The four soils were pretreated(i.e.,soaked with 0.01 mol L-1 K2SO4)or not before GRSO4 suspension was added.Bars are standard errors(n=3).
Effect of Cr(VI)b/Fe(II)s mole ratio on Cr(VI)stabilization efficiency
Figure 6 shows the residual concentrations of Cr(VI)(extracted by phosphate buffer)in soil samples after reaction with GRSO4at various Cr(VI)b/Fe(II)smolar ratios.Notably,Cr(VI)stabilization efficiency increased with the decreasing Cr(VI)b/Fe(II)smole ratio.In all studied soil samples,the maximum stabilization efficiency was obtained at Cr(VI)b/Fe(II)sratio of 0.20.At the highest Cr(VI)b/Fe(II)sratio(0.46),where the availability of Cr(VI)for GR was not determinative,the Cr(VI)stabilization efficiency in acid soils was higher than that in alkaline soils.This is consistent with the effect of proton concentration on Cr(VI)reduction.Analysis of Cr(III)in soil solution after reaction with GRSO4revealed that all reduced Cr(III)was precipitated as Cr/Fe/OH oxyhydroxide or adsorbed by soil components.
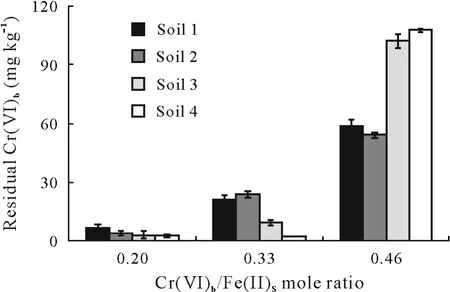
Fig.6 Residual phosphate buffer-extractable Cr(VI)(Cr(VI)b)of the four soils after reacting with hydroxysulfate green rust(GRSO4)for 60 min at soil/solution ratio of 0.1 and different initial Cr(VI)b/Fe(II)s mole ratios,where initial Cr(VI)b is the Cr(VI)b of the soils after four wetting-drying cycles and Fe(II)s is the amount of structural Fe(II)in the GRSO4 added.The four soils were pretreated(i.e.,soaked with 0.01 mol L-1 K2SO4)before the reaction.Bars are standard errors(n=3).
The initial and final pH values of the soil suspensions before and after reaction with GRSO4are presented in Table SI(see Supplementary Material for Table SI).The final pH values of the soil suspensions increased between 0.1 and 0.5 units due to the Cr(VI)reduction by GRSO4.
Effect of soil/solution ratio on Cr(VI)stabilization efficiency
The results showed that increasing soil/solution ratio caused a decrease in Cr(VI)stabilization efficiency(Fig.S2,see Supplementary Material for Fig.S2).The residual concentration of Cr(VI)in soil samples at soil/solution ratios of 0.5,0.2,0.1,0.05,and 0.02 was 15.6,10.6,10.8,2.9,and 2.7 mg kg-1,respectively.It is evident that the release of Cr(VI)from soil to the aqueous phase was affected by the concentration gradient and was low at high soil/solution ratios,resulting in diminished stabilization efficiency.
Back-transformation of produced Cr(III)
The results for back-transformation of the produced Cr(III)in the batch system are illustrated in Fig.7a.The results indicated that the amount of chromium oxidized was proportional to the concentration of manganese oxides in soil and that chromium oxidation was a relatively slow reaction.Once birnessite was added to the soil,5.2% of the total initial Cr(III)was converted to Cr(VI)after two months.Soil 4 contained 84.5 mg kg-1easily reducible manganese,which was able to oxidize 2.1% of the initial Cr(III)to Cr(VI)(control sample 1).However,in the sample without GRSO4and with the addition of birnessite(control sample 2),no oxidation was observed.This indicates that the Cr(VI)producedviaoxidation was mainly from the Cr(III)in the Cr/Fe/OH oxyhydroxide structure.The results of experiments carried out under FC conditions are shown in Fig.7b.The results show that less than 0.05%of the initial Cr(III)was oxidized to Cr(VI)after six months.More contact between solid-Cr(III)and birnessite in the batch system,as compared with FC moisture condition,was responsible for the more oxidation of Cr(III).
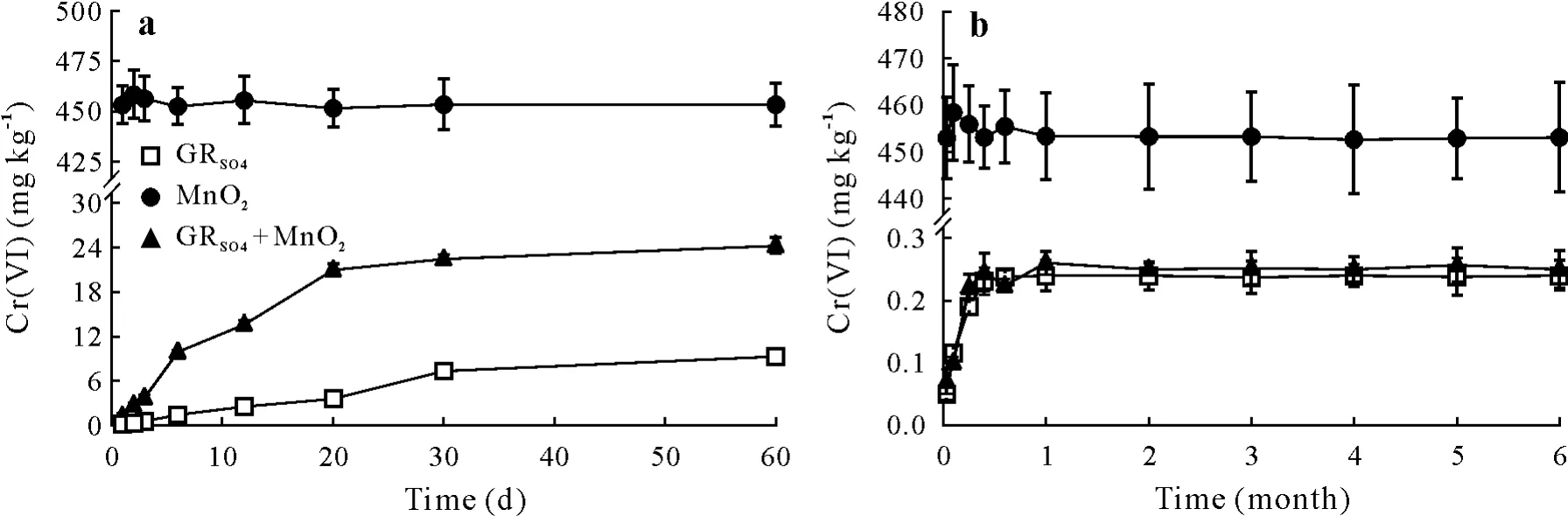
Fig.7 Concentrations of Cr(VI)produced in soil 4 with MnO2 or hydroxysulfate green rust(GRSO4)or both present in a batch system(a)or under field capacity moisture conditions(b).Bars are standard errors(n=3).
The dissolved Cr(III)can be readily oxidized to Cr(VI)by manganese oxides,but this process is limited when Cr(III)is precipitated as Cr(OH)3(Eary and Rai,1987).However,Bartlett and James(1979)showed that 13%of the freshly precipitated Cr(OH)3could be oxidized to dissolved Cr(VI)in the presence of manganese oxide.This has led to concern about the fate of the Cr(III)produced from the reduction of Cr(VI)by Fe(II)in the structure of GRSO4.
Analysis of reaction final product
Chemical analysis of the filtrate and solid precipitates at the end of the reaction between Cr(VI)and GRSO4confirmed that all Fe(II)in the system was oxidized to Fe(III)during the reduction and precipitated as the final end product.Measurement of Cr(III)concentration in solution also revealed that all reduced Cr was removed from solution by precipitation.To determine the chemical composition of the final reaction products and valences of the elements present,XPS analysis was performed.Based on whole region scan of the precipitates,the main elements at the surface were found to be C,oxygen(O),Fe,and Cr.The C 1speak region(285.0 eV)was used for calibration of the binding energy(BE)of subsequent scans.Figure 8 shows core-level spectra of the regions of Cr 2p,Fe 2p,and O 1s.The O 1sspectrum could be fit using three peaks at 533.4,532.2,and 530.6 eV,corresponding to O present in H2O,hydroxyl,and oxide species,respectively.These species are commonly observed in metal(oxy)hydroxides(Manninget al.,2007).Cr 2p1/2 and Cr 2p3/2 XPS line scans at 577.48 and 587.42 eV are attributed to Cr(III)(oxy)hydroxides.The magnitude of the spin–orbit splitting between the Cr 2p1/2 and Cr 2p3/2 peaks was 9.94 eV,which is a characteristic value for Cr(III)compounds(Ikemotoet al.,1976).The Fe 2ppeak at 712.2 eV corresponds to Fe(III)oxyhydroxide compounds(Yamashita and Hayes,2008;Hassandoostet al.,2019).According to the results of the XRD analysis(Fig.S3,see Supplementary Material for Fig.S3),the final product of the reaction was amorphous,which is consistent with findings of other researchers(Rubyet al.,2003).
From these findings,it can be concluded that removal of Cr(VI)by GRSO4occurs through the reduction of Cr(VI)to Cr(III)by Fe(II)sfollowed by incorporation of the produced Cr(OH)3with Fe(III)oxyhydroxides.The structure of GRSO4and the mechanism of Cr(VI)removal are presented schematically in Fig.9.First,adsorption of Cr(VI)ionson the outer surfaces of GRSO4occurs simultaneous with their exchange with interlayer sulfate anions.Then,Cr(VI)is rapidly reduced to Cr(III)by Fe(II)s,leading to gradual disordering of the GRSO4structure.Finally,reddish-brown precipitates of Cr(III)/Fe(III)oxyhydroxides form.Although the reaction process and the final product in soils would not be exactly the same as those in aqueous solutions,due to the complexity of the soil system,analysis of the final product of the reaction performed in aqueous solution confirmed the general pathway of the reaction.

Fig.8 X-ray photoelectron spectroscopy(XPS)core-level spectra of the regions of the main elements found in the final product of the reaction between chromate and hydroxysulfate green rust(GRSO4).Points depict empirical data,and lines show the fitted curves by Gaussian-Lorentzian function.
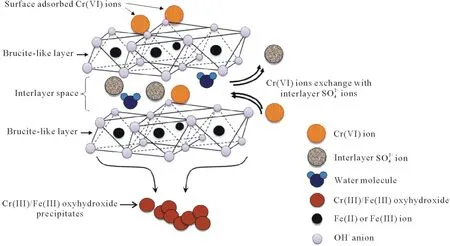
Fig.9 Simplified schematic showing the structure of hydroxysulfate green rust(GRSO4,Fe(II)4Fe(III)2(OH)12SO4·8H2O)and the mechanism of Cr(VI)reduction by GRSO4.
CONCLUSIONS
The reductive stabilization of Cr(VI)using GRSO4was studied in four Cr(VI)-spiked soil samples.Subsequently,the effects of various experimental conditions on the stabilization efficiency were investigated.The results indicated that freshly synthesized suspension of GRSO4was highly efficient and rapid in stabilizing Cr(VI)in Cr(VI)-spiked soils by reducing Cr(VI)to Cr(III).The extent of Cr(VI)stabilization by GRSO4was dependent on the Cr(VI)b/Fe(II)smole ratio and the rate of Cr(VI)release from soil to the aqueous phase during the reaction.The latter was influenced by soil pretreatment process,soil/solution ratio,and,particularly,the chemical properties of soils.The stabilization efficiency in alkaline soils with low OC content was higher than that in acid soils containing high OC,due to the higher rate of Cr(VI)release from alkaline soils.When Cr(VI)was better retained by soil,the rate of Cr(VI)release was lower than that of Cr(VI)reduction by GRSO4.In such systems,the rapid oxidation of unreacted GRSO4by dissolved oxygen before the complete release of Cr(VI)led to a decrease in Cr(VI)stabilization efficiency.Once the oxidizing condition was established in GRSO4-amended soil,less than 0.22 mg Cr(III)out of 492 mg Cr(III)per kg of soil was oxidized to Cr(VI)under FC moisture conditions over a long-term incubation period(6 months),confirming the high stability of the produced Cr(III)precipitate against oxidation.Further studies are needed to demonstrate if GRSO4would be successful for the remediation of Cr(VI)-contaminated soils under field conditions.
ACKNOWLEDGEMENT
The authors sincerely thank the University of Tabriz,Iran for providing support.
SUPPLEMENTARY MATERIAL
Supplementary material for this article can be found in the online version.
杂志排行

Pedosphere的其它文章
- News First Hot Paper among the papers published in Pedosphere
- Soil property mapping by combining spatial distance information into the Soil Land Inference Model(SoLIM)
- Toxicity of lead pollution to the collembolan Folsomia candida in Ferri-Udic Cambosols
- Proximal sensor-enhanced soil mapping in complex soil-landscape areas of Brazil
- Effects of porous clay ceramic rates on aeration porosity characteristics in a structurally degraded soil under greenhouse vegetable production
- Adsorption of emerging sodium p-perfluorous nonenoxybenzene sulfonate(OBS)onto soils:Kinetics,isotherms and mechanisms