面向新一代测序数据的病原微生物检测算法
2021-10-14袁细国
李 杰,李 苗,袁细国
1.西安翻译学院 商学院,西安 710105
2.西安电子科技大学 计算机科学与技术学院,西安 710071
微生物组学研究是生物信息学领域的热点之一,病原微生物在物种水平上的检测,对于交叉感染疾病和传染疾病的有效防御和精准诊疗具有十分重要的意义和价值。在传统检测方法中,往往是通过纯培养法培养微生物的方式进行观察和鉴定,但由于可培养的微生物物种极其有限,绝大多数微生物在现有实验和技术条件下难以培养,这极大地限制了病原微生物的检测能力。近些年来,随着新一代测序技术(Next Generation Sequencing,NGS)的快速发展,大批量且分辨率极高的基因组数据为微生物的分析提供了新的机会[1],为从DNA 分子水平上对病原微生物的检测提供了可靠平台。从基因组序列角度上看,微生物在物种水平上具有高度相似性,不同微生物物种的差异性碱基占比通常不到全基因组序列碱基数的百分之五,甚至只有几十个差异性碱基,这使得微生物物种的鉴别具有较大挑战性[2]。
微生物组学的研究对象主要是细菌,细菌rRNA按沉降系数分为3 种:5S、16S 和23S rRNA。16S rDNA(简称16S)[3-4]是细菌染色体上编码rRNA相对应的DNA序列,存在于所有细菌染色体基因中。从分子水平上分类和鉴定微生物的基本思路是对16S rDNA[2,5]序列进行分析,这是因为16S rDNA 既具有保守性区域,又具有高变性区域[6],可变区序列因细菌不同而异,保守区序列基本保守,使其具备精确刻画微生物种类和遗传多样性的天然条件[7-10]。16S rDNA序列的长度为1 500个碱基左右,大小适中,既能体现不同菌属之间的差异,又能利用测序技术较容易地得到其序列。本文均在数量轻小的16S上进行探索,对样本中物种类型做出鉴定。
早期,基于物种识别的方法使用BLAST(Basic Local Alignment Search Tool)工具[11]将测序读段局部比对到参考序列,并计算比对的相似性分数。将测序读段分配到相似性分数最高的物种下,直至测序读段全部分配完毕,物种下有测序读段则样本中含有该物种。但BLAST 的计算量较大,所以很多以BLAST 为雏形的算法很难适应参考数据库规模扩大或数据测序深度增大的情形。另一种物种识别的方法,是基于样本的共享系统发育。使用最大似然估计[12]、贝叶斯后验概率[13]或邻域连接[14]的系统发育方法都已经开发出来。用系统发育的方法来表示生物体之间的关系是很清晰的,但是这些方法计算量很大。此外,虽然这些方法能够进行准确的分类,但系统发育方法往往灵敏度较低[15]。伪比对是一种基于k-mer[16-18]的快速算法,它使用参考数据库的de Bruijn图来识别查询序列的潜在匹配,而不将序列与参考序列对齐。Kallisto[19]是一种基于序列伪比对的物种成分识别算法,通过提取测序读段间共享的k-mers构建de Bruijn 图,计算测序读段来自于某个特定物种的可能性,进而判断样本中的物种成分。但构建de Bruijn图会占据巨大的存储空间。Karp[7]利用了伪对齐的速度和低内存要求,并采用EM 算法,该算法使用基本质量分数快速准确地分类微生物组样本。但是Karp在使用的时候需要输入很多相关文件,非专业人员使用起来相当困难。近年来,Lindgreen 等人[20]和Sczyrba等人[21]提出了宏基因组学中物种检测方法的性能评估指标。
针对传统培养方法的不足以及现有基于测序数据方法的缺点,本文提出一种面向新一代测序数据的病原微生物检测算法(NBMicro),将病原微生物样本测序数据比对到参考基因组中,过滤掉比对不成功的测序读段,针对比对成功的读段状态,提取三种有效特征,建立基于朴素贝叶斯分类算法,以对微生物物种存在性进行分类(即存在与不存在两类)。本文采用的朴素贝叶斯分类器,其算法逻辑简单、分类过程中时间复杂度和空间复杂度都较小,且分类效果较好。相比于其他方法,NBMicro 从序列比对结果中提取的3 种特征,能够很好地刻画微生物序列与参考基因组序列的吻合程度,从而有利于判断微生物物种是否存在。同时,在数据量较大的情况下,NBMicro 的计算时间也较短,操作简单。
1 研究方法
NBMicro 方法的基本框架如图1 所示,主要步骤包括数据预处理、特征提取和利用朴素贝叶斯判别物种存在性。首先将数据进行数据预处理,数据预处理是采用BWA 软件[22]将待测样本比对到参考基因组序列(reference,ref)中,将比对不成功的测序读段(read)去除。其次将预处理之后的数据进行特征提取,本文提取物种下比对结果的3个特征:read depth(读深)、gapscore、gradmean。read depth 是指ref 中某一片段中比对到的平均位点数;gapscore 是指ref 中未比对到的片段长度和;gradmean是指将ref下比对结果的read count图谱的变化特征量化,read count是指ref上每个位点上所比对的read数量。最后,将提取到的特征使用朴素贝叶斯分类器来判别物种存在性。由于提取到的3 个特征并非在同一标准下,所以先要将3个特征标准化使其在同一标准下,然后利用朴素贝叶斯分类器判别物种是否存在,将存在的物种输出。
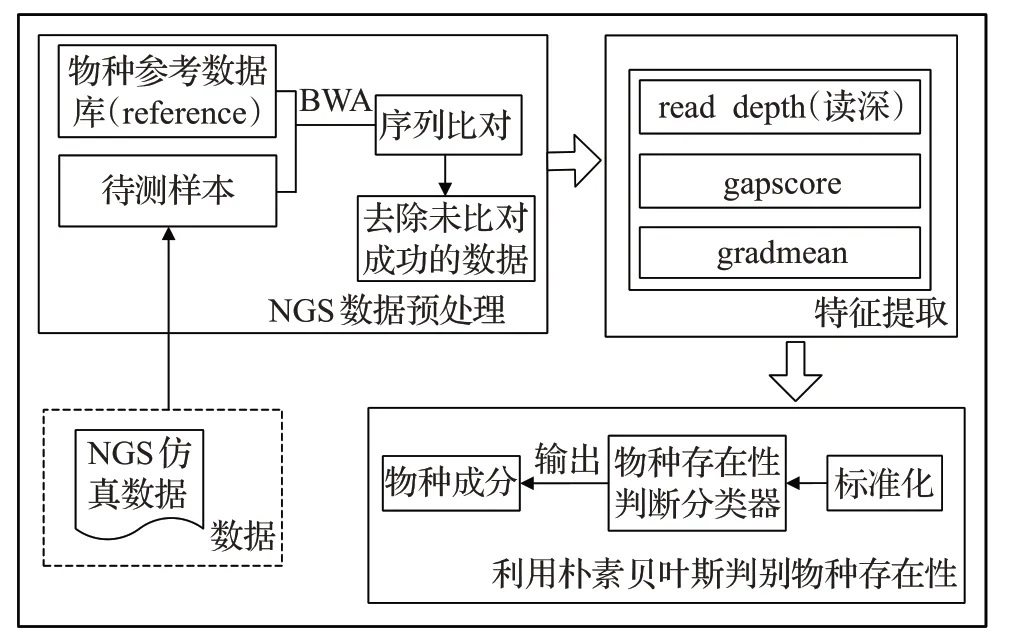
图1 NBMicro算法流程图Fig.1 Flow chart of NBMicro algorithm
1.1 特征提取
本文提取了3 种特征:读深(read depth)、空隙分数(gapscore)、梯度均值(gradmean),其详细说明如下所示。
(1)读深(read depth):是指ref 中某一片段中比对到的read 的位点数量除以片段长度,read depth 定义如公式(1)所示。样本组织中如果是含有该物种,那么在测序时该物种的DNA 序列往往会被采集到,从而获得相应的测序读段。从观察到的测序数据角度上看,若某微生物物种参考基因组比对了一定数量的读段(read),就恰好为该物种存在提供了有利证据。从物种下比对结果的read depth而言,read depth越高,则该物种比对到的的read越多,该物种存在的可能性越大。

(2)空隙分数(gapscore):是指将ref 下比对结果的gap 和,其中gap 指ref 中未必对到的片段。若物种比对后得到的gap 集合为G={g1,g2,…,gn},gi表示第i个gap 的长度,则物种比对结果下产生gap 的得分如公式(2)所示。将测序数据比对到参考基因组之后,参考基因组中有的位点比对到了读段,有的没有比对到,比对到的位点越多,则该物种比对的效果越好。gap 得分将所有未比对到的位点数统计起来,可以体现物种比对的效果,从而体现该物种存在的可能性。

如图2所示是一个ref比对结果的read count图谱,图中一共有7个gap,将g1,g2,…,g7加起来作为物种比对的gap 得分。该物种比对之后发生的gap 得分越小,说明比对的效果越好,该物种的存在性越大,反之亦然。
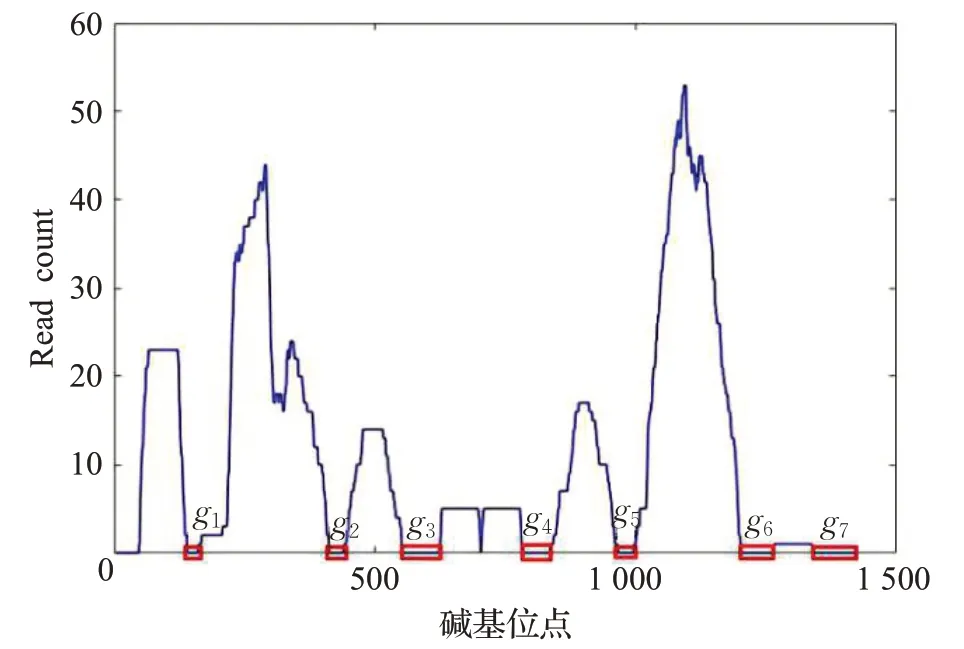
图2 read count图谱Fig.2 Read count profile
(3)梯度均值(gradmean):是指将ref中比对结果的read count 图谱做傅里叶变换,然后计算傅里叶频谱图上的峰值变化的梯度,并将梯度平均。其中read count是指参考序列上每个位点比对到读段的数量,随着参考序列位点变化的read count可用read count图谱表示出来;傅里叶变换是指将ref的位点分布函数变换为ref的频率分布函数;傅里叶频谱图上的峰值变化,即梯度的大小,其意义是ref 上某一点与邻域点差异的强弱。将测序数据比对到参考基因组之后,每一个参考序列都有一个随位点变化的read count图谱,将这个read count图谱进行傅里叶变换之后分析随频率变化的函数的峰值,峰值波动小,那么该峰值对应的位点与领域位点的差异较小,说明该物种比对效果较好,该物种存在的可能性较大。read count图谱的傅里叶变换如公式(3)所示:

式中,t表示碱基位置;N表示采样点数(即对碱基位点等间隔采样,采样最终得到的read count 点数为N个);u表示频率,u的取值范围为,其中fs表示采样频率,,T为周期,在这里取2π,u的取值为0,
图3 显示了一条ref 随着位点变化的read count 图谱,图4是图3进行傅里叶变换之后的结果,图4横轴表示碱基位点N点等间隔采样数,其中(N=100)。具体的采样操作为,将图3中的1 300个碱基位点,每隔13个碱基位点采集一次样本,一共采集了100 次。图4 纵轴是信号强度(即read count变换后的信号强度)。图4中的红线表示频谱变化,蓝色点标记的是峰值变化。假设相邻两个峰值为P1(v1,w1)和P2(v2,w2),梯度计算如公式(4)所示:

图3 ref随着位点变化的read count图谱Fig.3 Read count profile of ref as locus changes

图4 Read count傅里叶变换图像Fig.4 Read count Fourier transform graph

对于每一条ref 求出其随着位点变化的read count图谱的傅里叶变换,求出各个相邻峰值的梯度,再将所有的梯度求均值作为ref 下下比对结果的一个特征,其公式如(5)所示,其中M表示峰值的数量。

1.2 利用朴素贝叶斯判别物种存在性
基于上述提取的3个特征,采用朴素贝叶斯分类器对物种存在性进行判别。朴素贝叶斯是假设特征之间强独立下运用贝叶斯定理为基础的概率分类器。对于给出的待分类项,算出在此项出现的特征下各个类别出现的概率,将待分类项分到概率最大的类别下。朴素贝叶斯分类器有算法简单,并且对小规模数据分类效果好等特点。
微生物物种特征集合X⊆Rd是d维向量集合,类标记集合Y={c1,c2,…,ck},训练数据T={(x1,y1),(x2,y2),…,(xD,yD)} 是由P(X,Y) 独立同分布产生,其中是第i个样本的的第j个特征,j=1,2,…,d,yi∈{c1,c2,…,ck}表示第i个样本分类的取值可能有k个。
类别Y=ck的先验概率如公式(6)所示:

公中,I是指示函数,指微生物物种训练数据集合T中哪些类别取值为ck,I(yi=ck)表示如果第i个样本在类别ck中则为1,否则为0。由于公式(6)会出现概率为0的情况,是指ck中没有样本。会影响到后续的后验概率的计算,使分类产生偏差,本文采用贝叶斯估计解决这一问题,其定义公式如(7)所示:

条件概率P(Xj=x|Y=ck)表示Y=ck已发生的条件下事件Xj=x发生的概率,由于本文提取微生物物种特征都是连续分布的,本文使用高斯分布来表示连续特征的类条件概率分布,其定义如公式(8)所示:

式中,μ表示均值,σ2表示方差,μjk可用类ck的所有训练记录关于Xj的样本均值来估计,可用这些训练记录的样本方差来估计。
朴素贝叶斯法分类时,对于给定的输入x,通过学习到的模型计算后验概率分布P(Y=ck|X=x),将后验概率最大的类作为x的类输出,其定义如公式(9)所示:

假设特征条件独立,公式(9)中分母对于所有的ck都是相同的,所以可写为公式(10):

将后验概率最大的类作为x类的输出,如公式(11)所示:
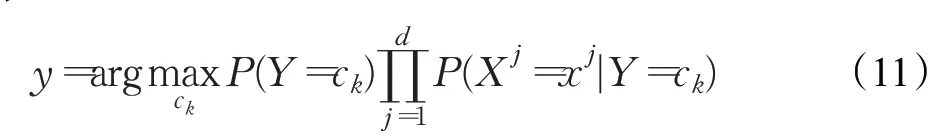
本文1.1 节提取了每个物种的3 种特征,生成特征向量(read depth、gapscore、gradmean),由于数据的不标准会给分类带来影响,所以这里对特性向量进行zscore 标准化。本文的分类只有两类,若该物种存在则该物种的类别(lable)记作1,否则记作0。对由此得到的数据集划分成8∶2的两个子样本,分别用作朴素贝叶斯分类器的训练集和验证集,对物种进行存在性判别,并将存在的物种输出。
2 仿真实验与分析
仿真实验对于算法性能的评估十分重要,本文采用ART软件[23]仿真数据,设计了11组小规模数据和5组较大规模的数据。
为了训练分类器来检测样本的中物种的成分,设计了11组小规模不同成分的样本。由于数据库中物种的相似度过高,匹配到相似物种的可能性较大,这里加入其他未知物种作为干扰。为了与真实样本受未知物种干扰的事实相一致,在仿真样本中添加了不同干扰程度的未知物种,干扰程度范围在[0,2.0],以0.2 为步长增长。每组样本的对应数据库有20 条物种,选取其中10条物种和不等量的干扰物种,并使用ART 软件生成仿真数据,仿真数据的覆盖度为800X,read长度为75。具体干扰程度如表1所示。
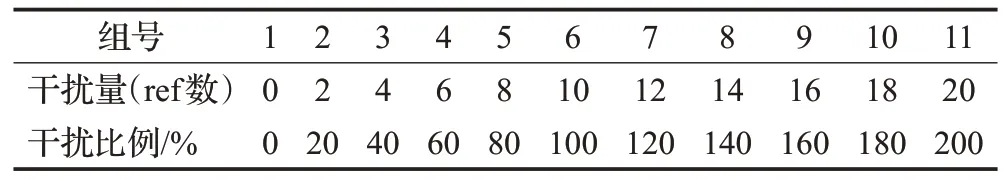
表1 仿真样本表Table 1 Simulation sample table
本文考虑了实际情况下人类测序读物(简称人类读物)的干扰,然后设计了5组样本,其中人类测序读物与微生物测序读物(简称微生物读物)的比例分别为0∶10、2∶8、4∶6、6∶4和8∶2。对于每个组,生成6个具有不同程度未知物种干扰的干扰样本的模拟样本,这些干扰样本的不相关物种干扰程度以步长0.2 在范围[0,1.0]中变化。每个样本的生成方法与11组小规模样本的相同。
本文选取3种指标评价成分检测的性能,分别是P(精度)、R(召回率)和F1-score 指标。F1-score 值越大,成分检测的性能越好。F1-score指标如公式(12):

图5是在不同干扰程度的样本上的P值和R值,选取了Karp、Kallisto方法与NBMicro方法做对比,不相关物种的干扰程度从0 逐步增加到2.0。图5 横坐标表示不同干扰程度,纵坐标表示样本的P值和R值,其中Karp_R表示Karp 方法的R值,Kallisto_R表示Kallisto方法的R值,NBMicro_R表示本文方法的R值,Karp_P表示Karp 方法的P值,Kallisto_P表示Kallisto 方法的P值,NBMicro_P表示本文方法的P值。从图5中可以看出,3种方法的R值都很高,说明但凡认定物种存在,则几乎是正确的。3 种方法中NBMicro 的P值较其他两种高,说明NBMicro将存在的物种识别的效率更高。
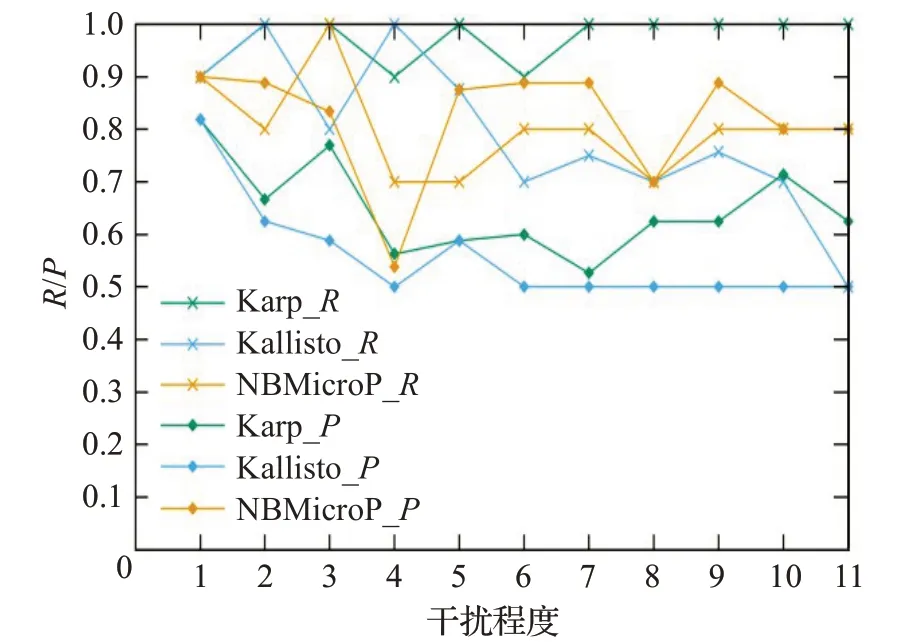
图5 11组样本上的R和PFig.5 R and P on 11 samples
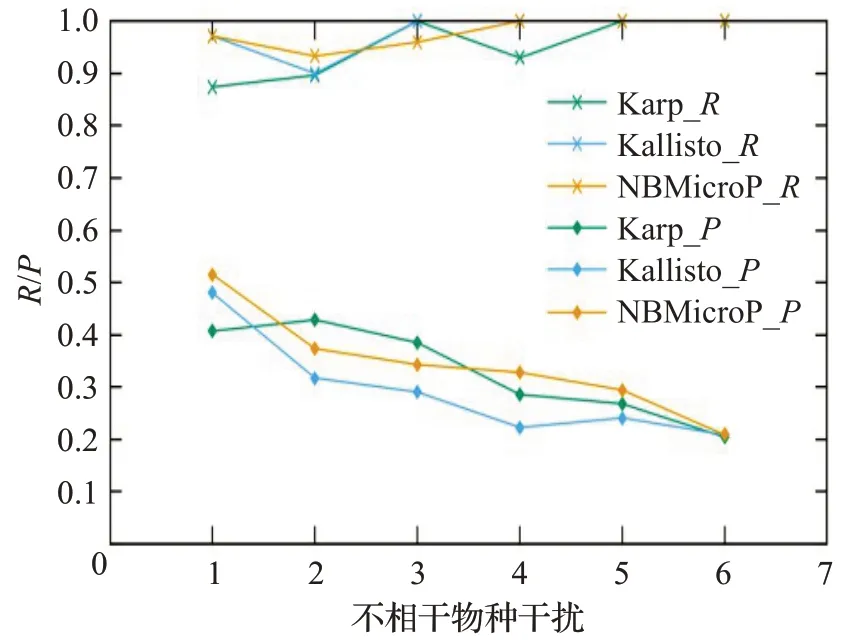
图6 人类读物与微生物读物比例0∶10的R和PFig.6 R and P when the ratio of human read to microbiological read is 0∶10

图7 人类读物与微生物读物比例2∶8的R和PFig.7 R and P when the ratio of human read to microbiological read is 2∶8
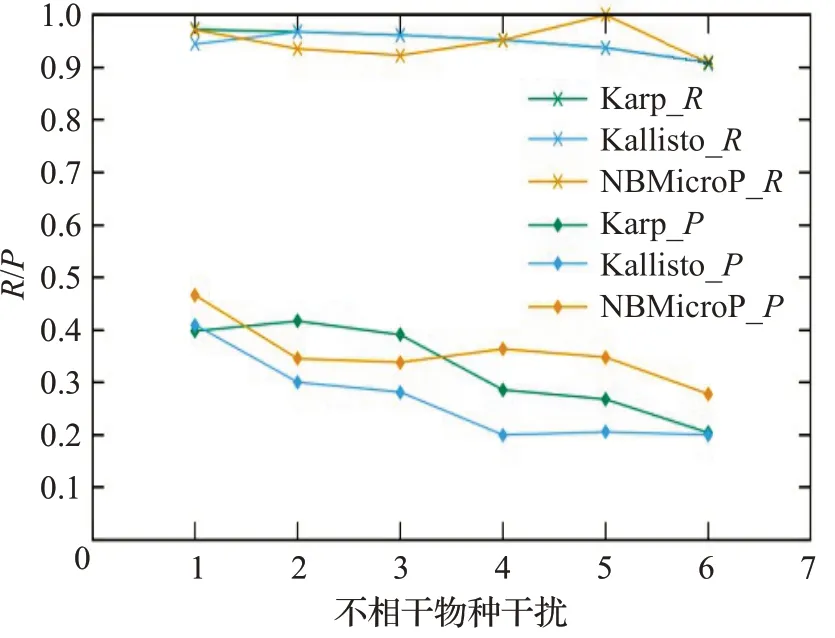
图8 人类读物与微生物读物比例4∶6的R和PFig.8 R and P when the ratio of human read to microbiological read is 4∶6
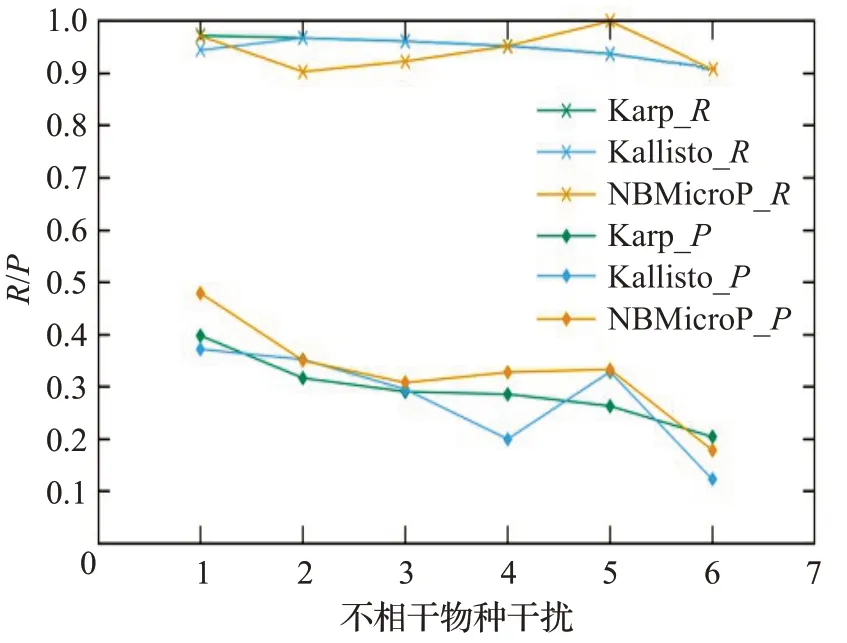
图9 人类读物与微生物读物比例6∶4的R和PFig.9 R and P when the ratio of human read to microbiological read is 6∶4
图6~10分别对应于5组较大规模样本的P值和R值,其中人类测序读数与微生物测序读数的比率分别为0∶10、2∶8、4∶6、6∶4 和8∶2。每个图都是确定了人类测序读数的比率,不相关物种的干扰程度从0逐步增加到1.0 时(从0.2 逐步增加到0.2)3 种方法的P值和R值。可以清楚地看到,3种方法的R值都较高,接近与1。在P值方面,NBMicro 方法总体上略高于其他两种方法。随着不相关物种的干扰程度的增加,所有方法的P值明显降低,说明随着不相干物种的增加,存在的物种的识别效率会有所影响。然而R值不随干扰程度变化而变化,说明算法识别出的物种的正确率不随干扰程度变化。此外,图6~10 的基本走势相似,说明P值和R值不受人类测序读物的干扰的影响。

图10 人类读物与微生物读物比例8∶2的R和PFig.10 R and P when the ratio of human read to microbiological read is 8∶2
图11是在不同干扰程度的样本上的F1-score值,本文选取了Karp、Kallisto 和Mothur[24]方法与NBMicro 方法做对比,不相关物种的干扰程度从0逐步增加到2.0。黑色线代表NBMicro 方法的F1-score 值,就整体而言NBMicro方法的物种识别度比其他3种方法高。

图11 11组样本上的F1-scoreFig.11 F1-score on 11 samples
图12~16 分别对应于5 组样本,其中人类测序读数与微生物测序读数的比率分别为0∶10、2∶8、4∶6、6∶4和8∶2。每个图都是确定了人类测序读数的比率,不相关物种的干扰程度从0 逐步增加到1.0 时(从0.2 逐步增加到0.2)4 种方法的F1-score。图12~16 中黑色的折线代表NBMicro 方法的F1-score 值,可以清楚地看到,NBMicro 方法在物种识别方面较其他方法有较高的准确性。此外,随着不相关物种的干扰程度的增加,所有方法的F1-score明显降低,说明随着干扰程度的增加物种识别的准确度不断降低,那么物种识别受到了干扰程度的影响。图12~16的基本走势不变,说明不受人类测序读数影响。
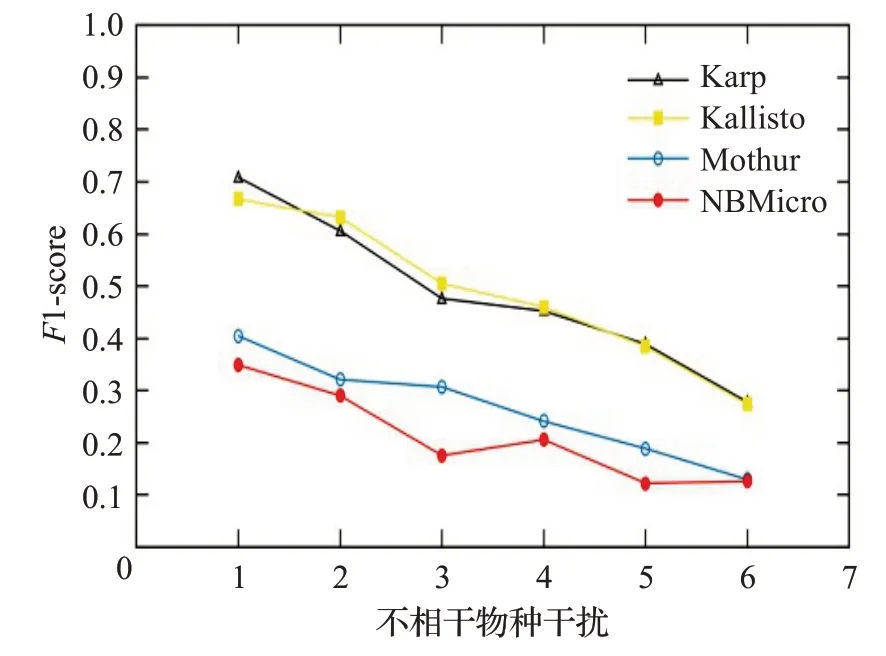
图12 人类读物与微生物读物比例0∶10时的F1-scoreFig.12 F1-score when the ratio of human read to microbiological read is 0∶10

图13 人类读物与微生物读物比例2∶8时的F1-scoreFig.13 F1-score when the ratio of human read to microbiological read is 2∶8

图14 人类读物与微生物读物比例4∶6时的F1-scoreFig.14 F1-score when the ratio of human read to microbiological read is 4∶6
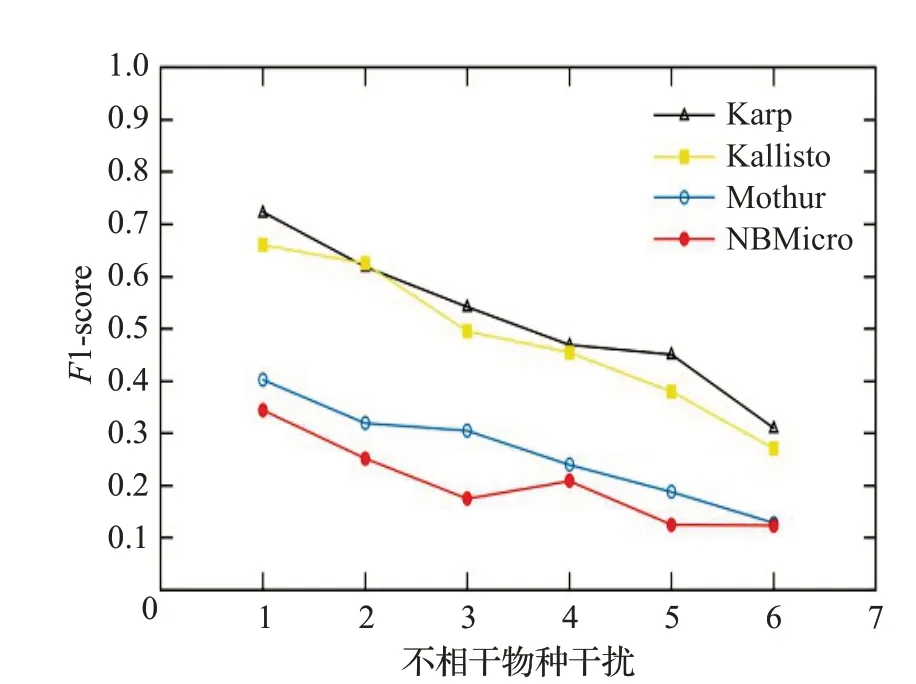
图15 人类读物与微生物读物比例6∶4时的F1-scoreFig.15 F1-score when the ratio of human read to microbiological read is 6∶4
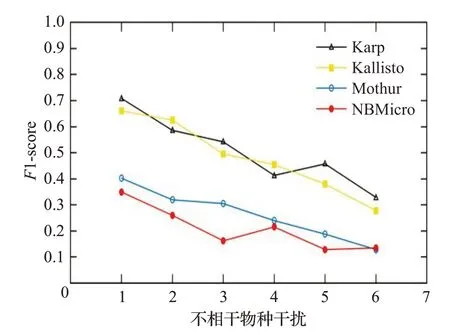
图16 人类读物与微生物读物比例8∶2时的F1-scoreFig.16 F1-score when the ratio of human read to microbiological read is 8∶2
从上述实验结果上看,本文方法在不同仿真情景下,都有较好的检测性能。其最主要的原因在于,所提方法不仅关注比对读段数,而且关注了比对读段在参考序列上的分布状态。由于读段通常较短,会存在多比对和错误比对情况,若仅仅关注比对读段数,会引起微生物物种判断的偏差,那么,比对读段的分布状态能够提供额外有用信息,这也促使本文方法在数据受到干扰情况下也能保持较好检测性能的原因。
为了验证本文提出的3种特征:read depth(读深)、gapscore 以及gradmean,对本构建的微生物检测算法的贡献度或重要性的差异,本文设计了一个实验。选取其中的两个特征在本文设计的11组小规模数据上进行检测,并且计算检测结果的P值、R值和F1-score 值,如果其检测结果较差,则意味着没被选的那个特征比较重要,反之亦然。如图17 是分别选择其中两个特征进行检测的结果,图中的“readdepth_R”是指使用了gapscore和gradmean 两个特征进行检测的结果的R值,图中的条形越高说明此特征越不重要。从图17 中可以看出gapscore 和gradmean 对本文的贡献较大,而read depth这一特征对本文方法的贡献度相对较小。

图17 readdepth、gapscore、gradmean重要程度对比Fig.17 Comparison of importance of readdepth,gapscore and gradmean
3 结论
本文提出一种面向新一代测序数据的病原微生物检测算法NBMicro,其思路是首先将测序数据进行预处理操作后,提取每个微生物物种参考基因组比对结果的3种特征,然后使用朴素贝叶斯分类器预测每个物种是否存在。通过仿真数据对所提算法进行验证,并与同行方法做了比较与分析,表明了所提算法的有效性和优势。因此,本文算法NBMicro 对于从DNA 分子数据上分析微生物物种具有推动作用。另外,本文还存在不足之处,比如微生物种群数据库规模有限,且仅考虑了微生物物种的检测,却没有考虑样本中每种微生物物种含量的估计,这在今后工作中将进一步完善。
