三七中皂苷类标准物质的规模化制备分离
2021-10-13王玥,丁燕,2,朱靖博,2
王 玥, 丁 燕,2 , 朱 靖 博,2
( 1.大连工业大学 食品学院, 辽宁 大连 116034;2.大连工业大学 植物资源化学与应用研究所, 辽宁 大连 116034 )
0 引 言
三七(Panaxnotoginseng(Burk.)F.H. Chen)具有活血化瘀、消肿定痛的功效[1-2],皂苷类物质是三七中主要的有效成分[3]。迄今已从三七的底根部、根基剪口、叶子、果实等部位分离和鉴定出120余种皂苷类化合物,基本阐明了他们的化学结构[4]。三七皂苷R1、人参皂苷Rg1、Re、Rb1、Rd为三七中含量较高的5种皂苷,质量分数占总皂苷的90%[5]。我国2015年版药典规定,以该5种皂苷质量分数为指标控制三七质量。有文献报道,人参皂苷Rb1、人参皂苷Rg1、三七皂苷R1对神经元损伤、血管内壁损伤都有明显保护作用[6-7]。因此,规模化制备三七中皂苷类标准物质对其药理作用的进一步研究与三七药材的质量控制具有重要意义。以往的研究中,多采用硅胶柱层析法[8]与大孔吸附树脂柱层析法[9]制备三七中皂苷类物质,存在单次上样量较少等影响制备效率的问题。LK1300S小颗粒树脂是一种新型的高分子吸附剂,具有吸附容量大、可重复利用等优点,对分离纯化中药中的有效成分具有制备效率高的优势。
近年来,关于三七中皂苷类物质制备分离的研究中,样品分离量普遍低于2 kg,且主要皂苷得率大多低于5%[10-12]。本研究首次采用LK1300S小颗粒树脂柱层析法与结晶法对三七总皂苷提取物进行规模化分离,并对其中含量较高的皂苷成分进行制备,以期得到纯度较高的人参皂苷Rb1组分。
1 材料与方法
1.1 材料与仪器
三七总皂苷提取物,珍宝岛药业股份有限公司;三七总皂苷对照品,大连博迈科技发展有限公司;薄层色谱硅胶,烟台市化学工业研究所;LK1300S小颗粒树脂,珍宝岛药业股份有限公司。
R502B旋转蒸发仪,巩义市予华仪器有限责任公司;DAC组合式动态轴向压缩工业色谱,大连博迈科技发展有限公司;高效液相色谱S6000,华谱新创新技术有限公司。
1.2 方 法
1.2.1 样品的制备
样品准备:取三七总皂苷提取物(呈棕色膏状)17 kg,加去离子水完全溶解。采用真空抽滤装置对样品水溶液进行过滤处理。
分析溶液的配制:取三七总皂苷对照品和提取物各50 mg,加甲醇溶解,配制成5.0 mg/mL的溶液。
1.2.2 填料预处理
取LK1300S小颗粒树脂20 kg于塑料桶内,加入乙醇没过树脂2~3 cm,浸泡3~4 h。用去离子水充分淋洗树脂至无明显乙醇气味。
1.2.3 装柱及平衡
采用动态轴向压缩工业色谱柱(DAC,250 mm×1 200 mm),将20 kg经预处理后的LK1300S小颗粒树脂缓慢填入色谱柱内,上下筛板为5 μm。柱内填料压实后,色谱柱的实际分离高度为1 070 mm。用150 L去离子水以体积流量300 mL/min对色谱柱内环境进行冲洗平衡。
1.2.4 分析方法
1.2.4.1 薄层色谱定性分析
展开剂为体积比6.5∶3.5的氯仿-甲醇混合液,香草醛-浓硫酸喷淋、电热炉105 ℃加热1~2 min,显色条件下对每组馏分进行薄层检测。根据Rf合并相似馏分。
1.2.4.2 液相色谱定量分析
色谱柱,C18(4.6 mm×250 mm,5 μm);流动相乙腈(A)-水(B),体积流量1.5 mL/min,检测波长203 nm,温度为室温,进样量10 μL。洗脱梯度:0~20 min,20% A; 20~45 min,46% A; 45~55 min,55% A; 55~60 min,55% A; 60~70 min,90% A; 72~90 min,20% A。
三七总皂苷对照品及提取物液相色谱如图1所示。图中三七皂苷R1保留时间16.2 min,人参皂苷Rg1保留时间23.3 min,人参皂苷Re保留时间27.4 min,人参皂苷Rb1保留时间37.4 min,人参皂苷Rd保留时间44.3 min。
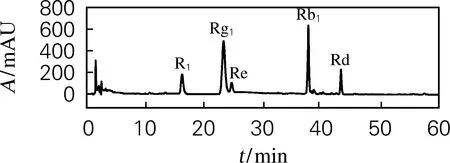
(a) 对照品
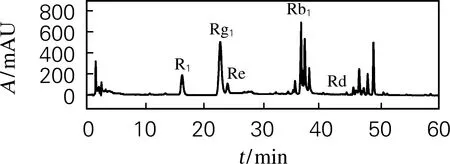
(b) 提取物
1.2.5 分析柱到制备色谱柱的线性放大
通过计算放大系数换算出上样量和体积流量,对制备色谱柱分离实验过程中条件进行优化[13-14]。P和A分别代表制备型和分析型色谱。
(1)
qP=qAS
(2)
QP=QAS
(3)
tP=tAVP/VAS
(4)
式中:S为放大系数,r为色谱柱半径,L为色谱柱柱长;q为上样量;Q为色谱体积流量;t为梯度延迟时间,V为柱体积。
线性放大只是一种理论依据,用于指导工业色谱制备条件的优化,而在实际生产时,为了提高效率,必然会出现过载的现象[15-17]。如表1所示,接下来实验过程中是在过载的情况下对样品进行制备分离。
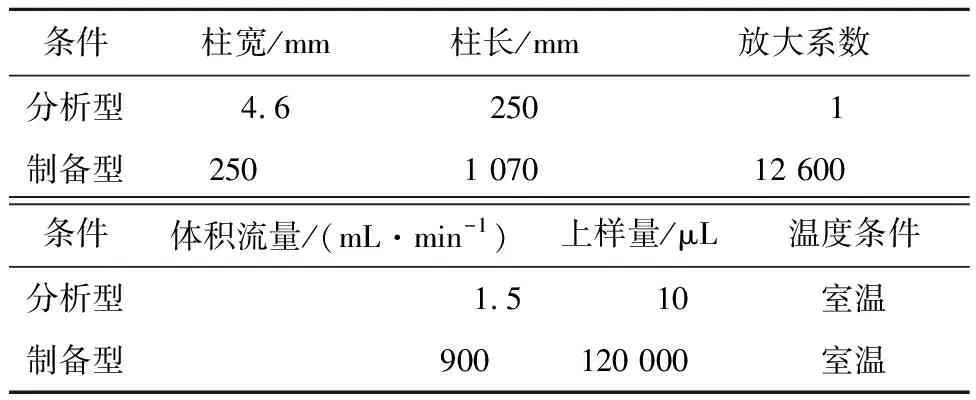
表1 分析柱到制备柱的放大设计参数Tab.1 Amplification design from analytical columnto preparative column
2 结果与讨论
2.1 LK1300S小颗粒树脂柱粗分离
将水溶样品滤液以100 mL/min体积流量进行上样。首先用250 L(1~10号)去离子水进行洗脱,收集流出液;再分别用体积分数为10%和70%的甲醇水溶液进行梯度洗脱,各洗脱250 L(11~20号、21~31号);最后用甲醇洗脱100 L(31~34号)。TLC跟踪分析,合并相似馏分,得到3组目标皂苷组分,如表2所示。从表2可以看出,经去离子水和10%甲醇水溶液洗脱得到非目标物质共1.8 kg,对样品进行除杂;从21号开始用70%的甲醇水溶液洗脱,洗脱液中逐渐出现目标皂苷物质,合并浓缩后经分析得到较纯的三七皂苷R1、人参皂苷Rg1、Re、Rb1富集组分,得率为37.05%。

表2 三七总皂苷提取物粗分离洗脱收集Tab.2 Crude separation, elution and collection of totalsaponin extracts of P. notoginseng
2.2 皂苷富集组分结晶除杂
向皂苷富集组分中加入30 L 30%的甲醇水溶液,室温静置一周后样品溶液中析出白色结晶,进行过滤处理。将所得母液组分浓缩称重,质量为5.63 kg;结晶组分挥干溶剂称重,质量为0.37 kg。对母液及结晶组分进行HPLC分析,结果如图2所示。图中母液组分中含原有4种目标皂苷成分;结晶组分中含多种非目标成分。说明通过本次结晶分离对皂苷富集组分进行了除杂,总纯度由48.8%提升至63.7%。

图2 母液及结晶组分的高效液相色谱图Fig.2 HPLC of mother liquor and crystallinecomponents
2.3 LK1300S小颗粒树脂柱二次制备分离
采用LK1300S小颗粒树脂柱对结晶除杂后所得皂苷富集组分进行二次分离。将水溶富集组分以100 mL/min上样,首先用20%和30%的甲醇水溶液各洗脱250 L(1~20号);再用50%和80%的甲醇水溶液各洗脱350 L(21~48号);最后用甲醇洗脱150 L(49~54号)。TLC跟踪分析,合并相似馏分,共得到4组目标皂苷组分,如表3所示。从表3中可以看出,经20%和30%的甲醇水溶液洗脱,得到非目标物质共0.1 kg,去除富集组分中的前杂物质;从第21号开始用50%的甲醇水溶液洗脱,洗脱液中逐渐出现目标皂苷物质;从第35号开始用80%甲醇水溶液洗脱,直至洗脱到第40号出现人参皂苷Rb1单物质,合并浓缩后质量为1.1 kg。如图3所示,经HPLC分析得到纯度为83.2%的人参皂苷Rb1组分,得率为6.47%。
3 结 论
采用LK1300S小颗粒树脂柱层析法结合结晶法对17 kg三七总皂苷提取物进行制备分离,最终得到1.1 kg纯度为83.2%的人参皂苷Rb1组分,得率提升至6.47%。该分离方法能有效地实现三七中人参皂苷Rb1的规模化制备分离。
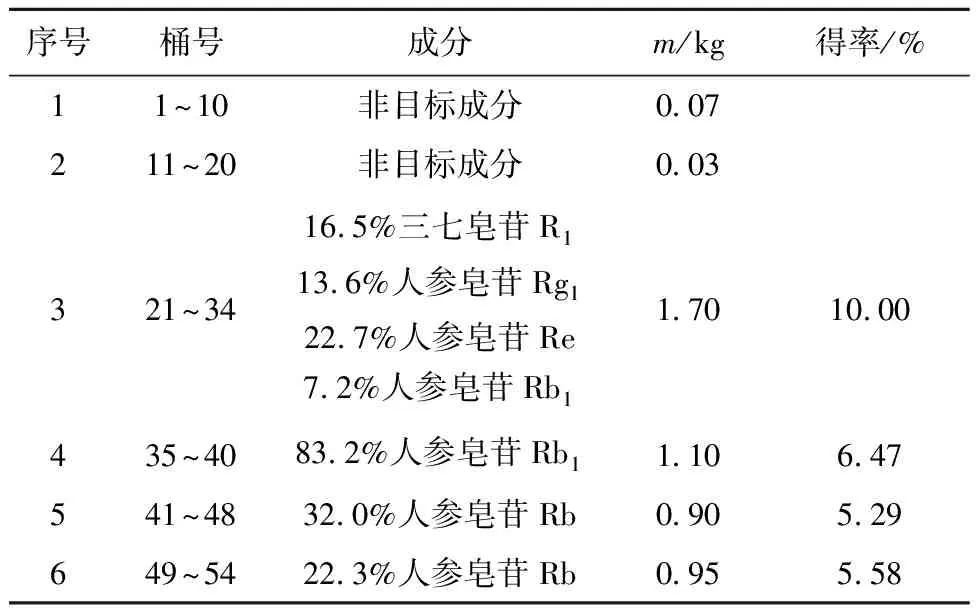
表3 皂苷富集组分柱层析分离洗脱采集表
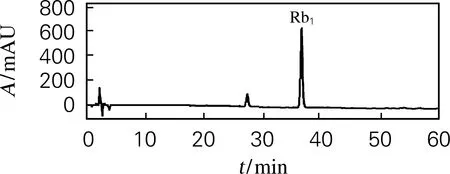
图3 人参皂苷Rb1组分的高效液相色谱图Fig.3 HPLC of ginsenoside Rb1
