丝状真菌代谢组学研究方法进展
2021-10-12闫震陈万权张昊刘太国刘长仲冯洁
闫震 陈万权 张昊 刘太国 刘长仲 冯洁

摘要 代谢组学研究的是生物体系受到内在和外在因素刺激产生的内源性代谢变化。近年来,随着代谢组学在病原真菌致病机理、真菌与植物互作以及新型生物活性物质开发研究领域的应用,真菌代谢组学越来越受到国内外的重视,同时也取得了较大的进展。由于丝状真菌类型和研究目的的差异,其研究方法不尽相同。本文主要综述近年来国内外丝状真菌代谢组学分析方法的研究进展,从丝状真菌样品制备、代谢物鉴定、数据分析、生物标志物发掘、代谢通路方面进行概述,以期为该领域研究者提供参考,进一步推进代谢组学在真菌研究领域中的应用。
关键词 丝状真菌; 代谢组学; 样品处理; 数据分析; 代谢通路
中图分类号: Q 591.1
文献标识码: A
DOI: 10.16688/j.zwbh.2020357
Advances in research methods of filamentous fungal metabolomics
YAN Zhen1, 2, 3, CHEN Wanquan1, 2, 4*, ZHANG Hao2, 4*, LIU Taiguo2, 4, 5*, LIU Changzhong1, FENG Jie2
(1. College of Plant Protection, Gansu Agricultural University, Lanzhou 730070, China; 2. State Key Laboratory
for Biology of Plant Diseases and Insect Pests, Institute of Plant Protection, Chinese Academy of Agricultural
Sciences, Beijing 100193, China; 3. Institute of Pomology, Chinese Academy of Agricultural Sciences,
Xingcheng 125100, China; 4. National Agricultural Experimental Station for Plant Protection at Gangu, Ministry
of Agriculture and Rural Affairs, Tianshui 741200 China; 5. Key Laboratory of Biological Hazard Factors (Plant Source)
Control of Agricultural Products Quality and Safety, Ministry of Agriculture and Rural Affairs, Beijing 100193, China)
Abstract
Metabolomics studies the endogenous metabolic changes in biological systems stimulated by internal and external factors. In recent years, with the application of metabolomics in the pathogenesis of pathogenic fungi and interaction with plants, the development of novel biologically active substances, more and more attentions have been paid to the study of fungal metabolomics, and great progresses have been made in this area. Due to the differences in the types of filamentous fungi and the purposes of the research, the research methods were diverse. This review summarized the research progresses in the metabolomic analysis methods of filamentous fungi in recent years, including filamentous fungal sample preparation, metabolite identification, data analysis, biomarker discovery and metabolic pathways. It is aimed to provide a reference for the researchers in this field and further promote the application of metabolomics to fungal research.
Key words
filamentous fungus; metabolomics; sample preparation; data analysis; metabolic pathways
代謝组学是对某一生物或细胞在一特定生理时期内所有低分子量(小于1 000)代谢产物进行定性和定量分析的一门学科,主要包括糖类、脂质、核苷酸、氨基酸等初级代谢产物以及萜类、聚酮类、非核糖体多肽、生物碱类等次级代谢产物[1-2]。虽然经常被视为转录组学和蛋白质组学的补充技术,但代谢组学确实具有明显的优势[3-5]。 首先,作为基因转录和蛋白质组表达的下游产物,代谢组与生物系统表型直接相关,可以直接动态地反映出细胞的生理生化过程,能够更准确直接地反映生命体终端和表型信息[6]。其次,相对于转录组或蛋白质组,代谢组的变化被放大,通过结果直接突出代谢途径和不同调控过程。代谢组学是功能基因组学所采用的组学技术中最新的一种[7],已经成为生物学的一个新领域,有望加快对功能未知基因的功能分析。
目前,代谢组学已扩展到各個领域,例如临床、制药、环境、动物、植物和微生物等[8]。相比于动、植物代谢组学,真菌代谢组学起步较晚,在过去的十年中,由于植物病原真菌在微生物系统中的重要性,代谢组学技术已被广泛用于植物病原真菌的研究中[9],主要侧重于真菌分类[10-12]、致病机理[3,13]以及与植物互作过程中代谢物功能和代谢途径[14-16]。国内外真菌代谢研究相关领域文献发表数量见图1,主要侧重于代谢产物、生物活性、抑菌活性、结构鉴定和发酵等方面的研究。代谢组学可用于检测正常的遗传发育以及寄主刺激引起的代谢组特征变化,从全局角度反映出真菌的表型变化[17],也可用于获得植物在感染病原真菌后产生的小分子代谢物[18-19]。为了研究植物的免疫力,还使用源自植物病原真菌的激发子来诱导寄主植物的免疫反应,进行代谢研究。许多植物病原真菌,例如镰刀菌Fusarium和曲霉Aspergillus都可以在寄主细胞中产生毒素[20]。因此,代谢组学还可以很好地检测毒素诱导的植物代谢途径的变化。
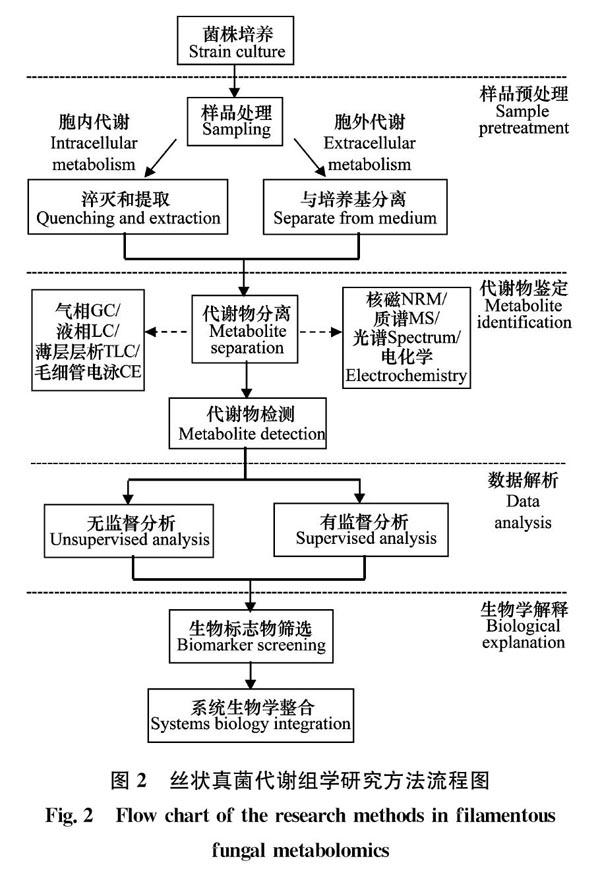
真菌代谢组分为胞内代谢物和胞外代谢物,通常将对胞内代谢物的定量定性分析称为代谢指纹分析(metabolic fingerprinting analysis),将胞外代谢物的定量定性分析称为代谢足迹分析(metabolic footprinting analysis)[21]。代谢指纹分析相比于代谢足迹分析,样品取样和准备过程较为复杂,但代谢指纹分析所涵盖的代谢物种类显著多于代谢足迹分析,所含信息更加丰富,所以当前以微生物为研究对象的代谢组学研究中,多数集中于代谢指纹分析。然而,由于真菌培养的样品中生物质浓度较低,限制了对细胞内数量少的代谢物的检测。以曲霉和青霉Penicillium为例,真菌细胞的次级代谢产物据估计达到10 000多种,但目前已知的代谢产物不足10%[22];另外,将细胞外和细胞内代谢物的分离与细胞内代谢的淬灭结合起来非常困难, 致使真菌代谢组学的研究面临很多挑战[23]。真菌代谢组学研究方法主要包括样品制备、代谢物鉴定、数据解析、生物标志物发掘、生物学解释等一系列关键步骤(图2),本文主要围绕这几方面展开讨论阐述。
1 样品制备
样品制备是代谢组学分析的关键步骤,对随后的数据采集和生物学解释具有重要的意义[24]。因此,应根据研究目的针对性地选择样品处理方法,真实客观地反映样品中代谢物的存在信息。理想的样品制备方法应简单易于重复、步骤少可变性最低、成本低适用于大样本量,且能够保持样品的完整性[25]。由于细胞对外界环境变化极其敏感,故代谢产物的含量及水平会随环境改变迅速发生变化[26]。因此,应尽快完成采样并终止细胞代谢(淬灭),以确保分析时样品的代谢组成分与取样时一致。真菌样品制备主要包括菌株培养、快速取样、代谢淬灭和代谢萃取[27]。通常取样和淬灭步骤结合在一起以缩短处理时间。
1.1 菌株培养
菌株需要在一定的可控生物反应器中培养。生物反应器中的培养基组分、温度、pH、光照以及溶解于其中的O2、CO2 等必须是确定和易于控制与检测的,这样才能获得标准和重现的培养条件。真菌多采用分批培养和连续培养方式。Chen等[28]和Palyzov等[29]使用PDA培养基培养真菌,从中提取代谢产物,但由于PDA培养基中含有具有生物活性的马铃薯,其不同批次之间营养成分难以达到统一标准,仅适用于少量样品统一处理,对于大量样本检测的重复性操作不可控制,因此,培养基成分适合选择化学合成的物质,便于控制培养条件的一致性[30-31]。
1.2 快速取样
快速取样不仅能够防止底物浓度发生巨大变化,而且有助于维持代谢物的稳定性。对于真菌最关键的问题是菌丝与生长培养基的有效分离,目前主要分离方式是冷离心或快速过滤[32]。细胞外代谢产物的取样和样品制备相对简单,一般离心或过滤后,取上清液或滤液直接淬灭或冷冻储存[24]。然而对于胞内代谢物, Francisca等[33]研究黑曲霉Aspergillus niger时发现,通过离心分离菌丝体是不切实际的,因为没有形成合适的菌丝团沉淀,尽管Seifar等[34]曾尝试离心后清洗菌丝沉淀,但试管中仍会存在大量的培养基或清洗液,导致细胞外代谢产物残留。与离心相比,过滤可以更有效地去除菌丝体中的细胞外代谢物[31,33]。Juliana等[35]研究尖孢镰刀菌F.oxysporum聚酮类代谢物时,采用先离心后真空过滤的方法,但暴露时间过长。Choi等[36]分析酿酒酵母Saccharomyces cerevisiae胞内代谢物时,比较了淬灭后离心和快速过滤对代谢物泄漏的影响,同样表明快速过滤能够很大程度上减少代谢物的损失。尽管过滤过程中可能会导致具有快速周转率代谢物(例如,磷酸化的中间体)的数量在重复样品中会有所不同[37],但可以考虑通过中心代谢产物的变化补偿不一致性[26]。比如,磷酸烯醇丙酮酸、NAD+/NADP+和谷氨酸等。所以迄今为止,快速过滤仍然是丝状真菌的最佳取样方法[30]。
1.3 代谢淬灭
理想的淬灭技术应实现瞬间淬灭,快速淬灭酶活力并且保持细胞或生物的完整性,避免造成胞内代谢物外流[27]。由于真菌细胞壁的结构不同,对渗透压的耐受程度以及膜通透性也存在差异,通常很难通过单一的提取方法提取全部胞内代谢物[38]。针对不同样品发展了各类淬灭试剂,液氮、高氯酸、冷甲醇(-40℃)、热乙醇、冷缓冲盐(PBS、Tricine、HEPES或碳酸铵)等[39]。也有报道用等渗盐水(09%m/V NaCl,0.5℃)淬灭能有效防止细胞膜损伤,从而避免不必要的代谢物损失。尽管也有研究者提出采用快速热休克来使新陈代谢失活,但这可能导致热不稳定代谢产物降解并增加细胞通透性[23,40]。使用60%甲醇(-40℃)的淬灭方法在各种真菌中应用最广泛[7,22,31]。但许多研究证明冷甲醇淬灭会破坏细胞完整性,影响细胞内代谢物的回收,从而导致代谢物水平的低估[41-43]。Zheng等[44]研究黑曲霉代谢物时,从综合淬灭效率、细胞损伤和代谢产物泄漏3个方面评估了3种冷甲醇淬灭方法(-20℃ 40%甲醇,-40℃ 60%甲醇和-40℃纯甲醇)与液氮的差异,发现与冷甲醇淬灭方法相比,液氮对细胞完整性破坏最小,产生的代谢物更稳定。当测定胞内代谢物时,为避免受到胞外代谢物的干扰,有时需要洗涤菌丝以去除细胞外基质。洗涤缓冲液应符合生理水平,采用比较温和的溶液,最大程度减少代谢物泄漏,一般是预冷的PBS、生理盐水或蒸馏水[30,45-46]。包括洗涤在内的整个过滤过程要求在30 s或45 s内完成[30,47]。
1.4 代谢萃取
代謝物的提取是样品前处理过程中又一重要步骤,对于非靶标代谢组学分析,其无偏向性和全覆盖率的内在要求,对代谢物的提取提出了较高的外在要求。理想的方法应尽可能提高代谢物的覆盖率和回收率,保持代谢物的物理化学性质稳定,减少代谢物损失[48-49]。首要步骤是细胞破碎,使细胞膜可渗透提取剂(例如有机溶剂),从而释放细胞内代谢产物[18]。随后进行代谢物的提取和样品浓缩。细胞破碎方法分机械破碎和非机械破碎。机械破碎方法主要有超声破碎细胞,微波辅助萃取,加压液体萃取,超临界流体萃取、手动研磨,球磨机等。非机械破碎包括酶促裂解、物理溶解和化学溶解。物理溶解常用的是冻融和加热法,由于样品中存在热不稳定化合物,加热法不太适用于全组分。化学溶解法主要是各类有机溶剂对细胞膜的破坏而使细胞内代谢物释放[39]。
目前,常用的提取剂有冷甲醇、热乙醇、乙腈、乙酸乙酯、二氯甲烷、高氯酸或碱以及氯仿-甲醇混合液等[27,33,50-51]。Karpe等[52]提取尖孢镰刀菌胞内代谢物时采用乙腈/水/乙酸混合液,虽然乙腈除蛋白的效果比甲醇和乙醇好,但乙腈对代谢物的提取效率最低,可重复的代谢特征较少[21]。美国Scripps研究所通过聚类分析发现,100%的甲醇或包含甲醇的混合提取溶剂可以实现高重复性检出代谢物。Smart等[26]使用冷甲醇水溶液提取与冻融循环结合,提高了细胞的渗透性,增强了代谢产物的化学提取。Tomas等[48]的研究表明使用甲醇和水混合液对镰刀菌代谢产物进行提取,能够兼顾高极性和低极性的化合物。由于真菌微生物样品内代谢物种类多,物理化学性质多元(极性和溶解性等),到目前为止,还未有一种提取体系可实现全覆盖提取代谢物,因此根据研究目的有针对性地选取提取剂至关重要[9]。
2 代谢物鉴定
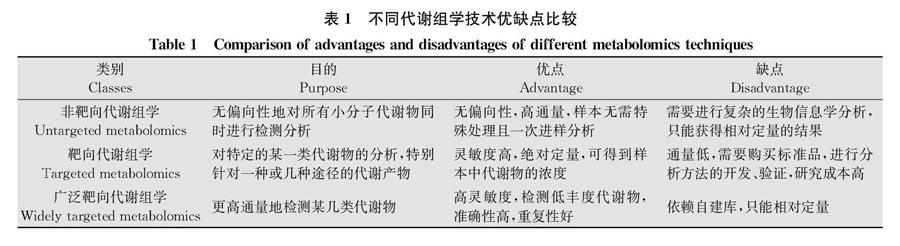
由于特定的代谢物在每一个含有它的有机体中都是相同的,所以代谢物鉴定技术是通用的。根据研究目的和研究技术不同,代谢组学主要分为非靶向代谢组学和靶向代谢组学两种典型的方法,前者可利用高分辨率质谱(high resolution mass spectrometry,HRMS)获得样品中尽可能多的代谢物信息,具有无偏性,但只能相对定量,通常主要用于区分代谢表型或发现差异代谢物的研究[21,53];后者则主要通过使用色谱(LC/GC)或串联低分辨率质谱(MS),针对特定代谢物进行分析,能够绝对定量,但覆盖率低。Chen等[54]基于Q-Trap质谱仪发展了“广泛靶向”代谢组学技术,将非靶向代谢组学高通量的优点与靶向代谢组学高准确度、高灵敏度的优点相结合,对关注的某类特定物质进行检测,生物标志物筛选更加准确,显著提高了信息覆盖率和定量准确性。三者的优缺点见表1。
代谢物分析鉴定的两大难点是生物体内小分子种类繁多、浓度相差悬殊,从真菌中获得代谢物化学成分的精确信息是一项具有挑战性的任务[55]。目前还没有一种代谢组学分析平台可以无偏向性地涵盖所有代谢物。色谱、质谱、核磁共振、红外光谱、紫外吸收、荧光散射、光散射等分离分析手段及其组合都出现在代谢组学的研究中[23],当前核磁共振(NMR)和质谱(MS)是代谢组学研究领域的主流分析平台,在代谢组学中发挥重要作用[56]。
2.1 核磁共振分析技术
NMR是一种无损、高通量的检测技术,利用生物流体的NMR光谱提供生物中所有小分子代谢物的丰富信息,并通过多变量统计分析和模式识别处理来确定相关生物的完整代谢图[9]。当前使用的NMR技术包括氢谱(1H-NMR),碳谱(13C-NMR)和磷谱(31P-NMR),其中最广泛使用的是1H-NMR[57]。NMR相对于MS的优势在于制备简单,物质的结构易于识别。此外,频谱上的信号强度与代谢物的浓度直接相关,可以准确定量代谢产物[58]。但是,NMR的灵敏度很低,很难同时检测生物体内浓度差异高的代谢物系统,并且无法区分具有非常相似结构和化学位移的代谢物,例如肌酸与磷酸肌酸,或尿苷二磷酸盐与尿苷三磷酸[3,17],限制了其在真菌代谢组学中的发展。
2.2 质谱分析技术
由于灵敏度高,质量准确度高和动态范围广,MS在代谢组学研究中起着越来越多的作用[59-61],因为它可以连接不同的色谱分离模式[62-64],例如气相色谱-质谱(GC-MS)、气相色谱串联质谱(GC-MS/MS)、液相色谱-质谱(LC-MS)、液相色谱串联质谱(LC-MS/MS)。气质联用主要针对具有挥发性且在高温下必须稳定的化合物。一些非挥发性代谢物(例如糖,氨基酸)可以通过衍生作用转化成挥发性化合物。尽管化学衍生化改善了代谢物的覆盖范围,但这可能为代谢组学研究引入了另一种变异性来源。该方法的最大优点是可以使用标准库进行结构鉴定,并且可以在大量数据库中检索[65]。与气质联用不同,液质联用主要利用“软电离”技术将分析物电离,通过选择离子监测模式(SIM)或多反应监测(MRM)模式进行,保留了完整的分子信息,这对于识别至关重要。由于它有更宽的线性范围,更好的可重复性和更高的灵敏度,被更加广泛地应用到靶向代谢组学研究中。
高分辨质谱(HRMS)凭借其普适性、高灵敏度和特异性的特点,逐渐成为非靶向和广靶代谢组学研究的主流技术[66]。目前主流的HRMS有飞行时间质谱(TOF-MS)、静电场轨道阱质谱(Orbitrap-MS)、傅立叶变换离子回旋共振质谱(FTICR-MS)[67]。相比TOF-MS,Orbitrap-MS具有更低的测量质量误差, 同时在仪器稳定性和重现性上显著优于TOF-MS, FTICR-MS结合了超高的质量分辨率和卓越的质量精度,是目前测量精度最高的质谱,但其扫描速度较前两者更慢, 在代谢组学研究中应用较少[21]。平行反应监测(PRM)是在HRMS平台上开发的新方法,对准确量化目标代谢物的贡献越来越大[61]。Zhou等[68]在Q-Exactive LC-MS系统上开发了基于PRM的目标代谢物检测策略,并进行了PRM和MRM测量的比较,结果表明,与QTRAP 6500上的MRM测量相比,PRM具有更高的质量精度和更全面的二级质谱信息。
不同分析工具各具优缺点,没有适合的单一的分析工具可以精确地鉴定和定量数千个感兴趣的小分子,需要组合使用不同的分析技术才能更好地覆盖具有广泛极性和分子量范围的代谢物[69]。
3 数据分析
色谱分离代谢物后,会生成大量光谱数据和多变量数据,频谱的每个信号峰都包含有关代谢物中各种物质的定性和定量信息[70]。因此,有必要使用统计学和化学计量学进行分析,具体分析流程见图3。首先需要对原始数据进行预处理,包括基线校正,特征检测,噪声过滤,峰提取,峰对齐,解卷积和归一化以消除干扰因素。可以使用软件来实现这些过程。例如MetAlign,MZmine,XCMS,METIDEA,AMDIS和MSFACTS等。许多仪器制造商还开发了自己的专有软件,例如MarkerLynx 和QI(美国Waters公司),AnalyzerPro(英国SpectralWorks公司),Mass Profiler(美国安捷伦公司),ChromsTof(美国Leco公司),MarkerView和SIEVE(美国Thermo公司)等[71]。
预处理后的数据需要进行多元统计分析和生物信息学分析,包括无监督分析和有监督分析两种[3]。无监督分析能够在未知样品信息的情况下将样品进行聚类分组,包括多变量数据分析(MADA)、主成分分析(PCA)、层次聚类分析(HCA)和自组织映射(SOA)[9,72]等。无监督分析方法不能忽略组内误差,如果样本组之间的差异太小或组内差异太大,则很难确定组之间的差异[73-74]。有监督分析包括偏最小二乘判别分析(PLS-DA)、正交偏最小二乘判别分析(OPLS-DA)、多元单变量数据分析(MUDA),线性判别分析(LDA)、神经网络(NN)和支持向量机分析(SVM)等[75]。有监督分析方法能够减少组内随机误差,突出组间系统误差[76]。通过这些多元变量统计分析,可以获得潜在的有效信息,并通过生物标志物和代谢途径找到差异代谢物[77],其中PLS-DA是使用最多的有监督分析方法,用于筛选有标记的代谢物[78]。
4 代谢通路分析
生物标志物确定以后,需结合所研究的问题考察其生物學功能。数据的可视化可以展示数据集的特性,通常绘制代谢网络的某一部分代谢途径图使数据可视化, 有助于理解代谢物之间的相互关系, 也能够与基因表达数据相联系进行功能基因组学研究,阐述病理生理作用相关的作用机制[79]。关于丝状真菌代谢通路研究相对较少,目前主要关注在中心碳代谢和能量转换方面。Liu等[51]探究了海藻糖生物合成基因TPS敲除导致禾谷镰刀菌F.graminearum基础代谢的变化,绘制了代谢途径图(图4),证明了TPS基因在糖酵解、TCA循环、磷酸戊糖途径中均具有调节作用。Vessela等[6]详细阐述了禾谷镰刀菌单端孢霉烯毒素的产生与其他中心碳代谢和次级代谢过程的关系,将收集的代谢物整合到代谢框架图中(图5),能够将可能发生在代谢途径之间的联系可视化,并可以指导与代谢轮廓修饰相关的生物学解释。Gu等[80]剖析了乙酰辅酶A合酶基因在炭疽病菌Colletotrichum higginsianum脂质代谢和毒力方面的作用,首次提出了参与病原体毒力的假设机制和模型(图6),揭示了脂肪酸β-氧化、肉碱穿梭、乙醛酸的生化途径,TCA循环、糖酵解、糖异生和脂肪酸生物合成之间的关联。Cao等[81]证明了转录因子CRF1在稻瘟病菌Pyricularia oryzae发育和致病性中的生物学功能,该因子参与脂肪分解,过氧化物酶体β-氧化、乙醛酸循环、糖异生和甘油代谢,影响附着胞的渗透性和毒力。Brisson等[82]研究了拟青霉Paecilomyces真菌柠檬酸与中心碳代谢之间的关系。Aguilar-Pontes等[83]描述了黑曲霉初级碳代谢网络途径,通过对高质量黑曲霉基因组的功能预测,深入挖掘了该途径中相关酶基因、反应及通路。然而,由于每种代谢物都可以参与多种生物合成途径[75],要正确解释代谢组学数据,绘制的代谢途径应该完整,即包含代谢物相关的所有反应。传统的代谢途径图,如常用的数据库KEGG,可能不包括所有的平行反应, 这也增加了代谢组学数据解释的难度。
Fig.6 Hypothetical model of the function of acetyl-CoA synthase in Colletotrichum higginsianum
5 展望
代谢组学是进行生物表型研究的主要手段之一,是系统生物学研究不可或缺的部分。真菌代谢组学是生物科学发展迅速的领域,随着样品制备方法的不断完善以及分析技术的快速发展,在真菌领域中将会发挥更大的作用。然而,与环境和人类代谢组学相反,其潜力尚未得到充分利用,从总体来看,它仍然处于发展阶段,目前代谢组学的最大挑战是如何克服从大量的代谢产物中找出特异性的生物标志物,获得最佳的生物学解释。另一个主要挑战是亚细胞区室化,因为代谢组学数据反映了各种细胞器中代谢物的总和。尽管从特定细胞器中分析代谢物是更可取的,但目前在保持这些结构代谢状态的同时分离细胞器是非常困难的。由于生物系统的复杂性,其他组学的配合和交叉至关重要。通过组合来自全基因组测序,基因表达谱,蛋白质组学和保守的生化途径信息,整合高通量、高分辨率的分析技术与生物信息学,实现自动分析和可视化,生成标准化代谢组学数据,进而构建细胞代谢模型。理想的模型可以预测生物体中存在的未知代谢途径,进而推进该领域的快速发展。
参考文献
[1] MATICH E K, NITA G, CHAVEZ S, et al. Applications of metabolomics in assessing ecological effects of emerging contaminants and pollutants on plants [J]. Journal of Hazardous Materials, 2019, 373: 527-535.
[2] 徐天潤, 刘心昱, 许国旺. 基于液相色谱-质谱联用技术的代谢组学分析方法研究进展[J]. 分析测试学报, 2020, 39(1): 10-18.
[3] 刘宏有, 陈柳龙, 高江涛. 代谢组及其在真菌研究中的应用[J]. 菌物学报, 2019, 38(12): 2078-2086.
[4] KELL D B, BROWN M, DAVEY H M, et al. Metabolic footprinting and systems biology: The medium is the message [J]. Nature Reviews Microbiology, 2005, 3(7): 557-565.
[5] HOLLYWOOD K, BRISON D R, GOODACRE R. Metabolomics: Current technologies and future trends [J]. Proteomics, 2006, 6(17): 4716-4723.
[6] VESSELA P, LAURIE L, STPHANE B, et al. Mycotoxin biosynthesis and central metabolism are two interlinked pathways in Fusarium graminearum, as demonstrated by the extensive metabolic changes induced by caffeic acid exposure [J/OL]. Applied and Environmental Microbiology, 2018, 84(8): e01705-17. DOI: 10.1128/AEM.01705-17.
[7] JOEL P A, GUMMER, CHRISTIAN K, et al. Metabolomics protocols for filamentous fungi [J]. Methods in Molecular Biology, 2012, 835: 237-254.
[8] GONG Zhigang, HU Jing, WU Xi, et al. The recent developments in sample preparation for mass spectrometry-based metabolomics [J]. Critical Reviews in Analytical Chemistry, 2017, 47(4): 325-331.
[9] CHEN Fangfang, MA Ruijing, CHEN Xiaolin. Advances of metabolomics in fungal pathogen-plant interactions [J/OL]. Metabolites, 2019, 9(8): 169. DOI: 10.3390/metabo9080169.
[10]KANG D, KIM J, CHOI J N, et al. Chemotaxonomy of Trichoderma spp. using mass spectrometry-based metabolite profiling [J]. Journal of Microbiology and Biotechnology, 2011, 21(1): 5-13.
[11]FRISVAD J C. Fungal chemotaxonomy, biosynthesis and molecular genetics of fungal secondary metabolites [M]. New York: Springer-Verlag, 2015, 2: 103-121.
[12]ALIFERIS K A, CUBETA M A, JABAJI S. Chemotaxonomy of fungi in the Rhizoctonia solani species complex performing GC/MS metabolite profiling [J]. Metabolomics, 2013, 9(1): 159-169.
[13]KONSTANTINOS A A, DENIS F, SUHA J. A metabolic profiling strategy for the dissection of plant defense against fungal pathogens [J/OL]. PLoS ONE, 2014, 9(11): e111930. DOI: 10.1371/journal.pone.0111930.
[14]TENENBOIM H, BROTMAN Y. Omic relief for the biotically stressed: metabolomics of plant biotic interactions [J]. Trends in Plant Science, 2016, 21(9): 781-791.
[15]ROJAS C M, SENTHIL-KUMAR M, TZIN V, et al. Regulation of primary plant metabolism during plant-pathogen interactions and its contribution to plant defense [J/OL]. Frontiers in Plant Science, 2014, 5: 17. DOI: 10.3389/fpls.2014.00017.
[16]TUGIZIMANA F, MHLONGO M I, PIATER L A, et al. Metabolomics in plant priming research: the way forward ? [J/OL]. International Journal of Molecular Sciences, 2018, 19(6): 1759. DOI: 10.3390/ijms19061759.
[17]NAKABAYASHI R, SAITO K. Integrated metabolomics for abiotic stress responses in plants [J]. Current Opinion in Plant Biology, 2015, 24: 10-16.
[18]FEUSSNER I, POLLE A. What the transcriptome does not tell-proteomics and metabolomics are closer to the plants patho-phenotype [J]. Current Opinion in Plant Biology, 2015, 26: 26-31.
[19]TAN K C, IPCHO S V, TRENGOVE R D, et al. Assessing the impact of transcriptomics, proteomics and metabolomics on fungal phytopathology [J]. Molecular Plant Pathology, 2009, 10(5): 703-715.
[20]NESIC K, IVANOVIC S, NESIC V. Fusarial toxins: secondary metabolites of Fusarium fungi [M]. Switzerland: Springer International Publishing, 2014, 228: 101-119.
[21]謝华里. 基于液质联用的黄曲霉代谢组学方法研究[D]. 北京: 中国农业科学院, 2017.
[22]靳梦琦, 李军, 朱凤妹, 等. 黑曲霉代谢组学研究进展[J]. 食品工程, 2017(4): 1-4.
[23]王智文, 马向辉, 陈洵, 等. 微生物代谢组学的研究方法与进展[J]. 化学进展, 2010, 22(1):163-172.
[24]MAGORZATA P, JULIA J, MICHA J M. Sample preparation procedures utilized in microbial metabolomics: an overview [J]. Journal of Chromatography B, 2017, 1043: 150-157.
[25]JULIJANA I, ELIZABETH J W. From samples to insights into metabolism: uncovering biologically relevant information in LC-HRMS metabolomics data [J/OL]. Metabolites, 2019, 9(12): 308. DOI: 10.3390/metabo9120308.
[26]SMART K F, AGGIO R B, VAN H J R, et al. Analytical platform for metabolome analysis of microbial cells using methyl chloroformate derivatization followed by gas chromatography-mass spectrometry [J]. Nature Protocols, 2010, 5(10): 1709-1729.
[27]席晓敏, 张和平. 微生物代谢组学研究及应用进展[J]. 食品科学, 2016, 37(11): 283-289.
[28]CHEN Fangfang, ZHANG Jingtao, SONG Xiushi, et al. Combined metabonomic and quantitative real-time PCR analyses reveal systems metabolic changes of Fusarium graminearum induced by Tri5 gene deletion [J]. Journal of Proteome Research, 2011, 10(5): 2273-2285.
[29]PALYZOV A, SVOBODOV K, SOKOLOV L, et al. Metabolic profiling of Fusarium oxysporum f.sp. conglutinans race 2 in dual cultures with biocontrol agents Bacillus amyloliquefaciens, Pseudomonas aeruginosa, and Ttrichoderma harzianum [J]. Folia Microbiologica, 2019, 64(6): 779-787.
[30]LU Hengqian, CHEN Haiqin, TANG Xin, et al. Evaluation of metabolome sample preparation and extraction methodologies for oleaginous filamentous fungi Mortierella alpine [J/OL]. Metabolomics, 2019, 15(4): 50. DOI: 10.1007/s11306-019-1506-5.
[31]RUTGER D D, LODEWIJK P J, CASPAR T H, et al. Intracellular metabolite determination in the presence of extracellular abundance: application to the penicillin biosynthesis pathway in Penicillium chrysogenum [J]. Biotechnology and Bioengineering, 2010, 107(1): 105-115.
[32]TUULIA H, SUSANNE W. Chromatographic methods in metabolomics [M]. Finland: Royal Society of Chemistry, 2013: 19-34.
[33]FRANCISCA L, JOSEPH J H, WALTER M G. Development of tools for quantitative intracellular metabolomics of Aspergillus niger chemost at cultures [J]. Metabolomics, 2015, 11(5): 1253-1264.
[34]SEIFAR R M, ZHAO Zheng, VAN DAM J, et al. Quantitative analysis of metabolites in complex biological samples using ion-pair reversed-phase liquid chromatography-isotope dilution tandem mass spectrometry [J]. Journal of Chromatography A, 2008, 1187(1/2): 103-110.
[35]JULIANA L, THOMAS P, LAURENT D, et al. Putative metabolic pathway for the bioproduction of bikaverin and intermediates thereof in the wild Fusarium oxysporum LCP531 strain [J/OL]. AMB Express, 2019, 9(1): 186. DOI: 10.1186/s13568-019-0912-4.
[36]CHOI J N, KIM J, JUNG W H, et al. Influence of iron regulation on the metabolome of Cryptococcus neoformans [J/OL]. PLoS ONE, 2012, 7(7): e41654. DOI: 10.1371/journal.pone.0041654.
[37]VILLAS B, SILSA G. BRUHEIM P. Cold glycerol-saline: the promising quenching solution for accurate intracellular metabolite analysis of microbial cells [J]. Analytical Biochemistry, 2007, 370(1): 87-97.
[38]李娟, 任路靜, 孙冠男, 等. 气相色谱-质谱联用技术及其在代谢组学中的应用[J]. 生物工程学报, 2013, 29(4): 434-446.
[39]谢华里, 李培武, 王秀嫔, 等. 非靶标代谢组研究温度对黄曲霉菌代谢的影响[J]. 分析测试学报, 2017, 36(11): 1304-1311.
[40]MATTHEW A L, CHARLES F B, ROBERT T K. Reducing time and increasing sensitivity in sample preparation for adherent mammalian cell metabolomics [J]. Analytical Chemistry, 2011, 83(9): 3406-3414.
[41]SOOAH K, DO Y L, GERT W, et al. Evaluation and optimization of metabolome sample preparation methods for Saccharomyces cerevisiae [J]. Analytical Chemistry, 2013, 85(4): 2169-2176.
[42]UDAYKUMAR K, SHIVAPPA H, AJJAMADA C K. Liquid chromatography and high-resolution mass spectrometry-based metabolomics to identify quantitative resistance-related metabolites and genes in wheat QTL-2DL against Fusarium head blight [J]. European Journal of Plant Pathology, 2018, 151(1): 125-139.
[43]ROHAN L, MLANIE J, GAIL C, et al. The induction of mycotoxins by trichothecene producing Fusarium species [J]. Plant Fungal Pathogens: Methods and Protocols, 2012, 835: 439-455.
[44]ZHENG Xiaomei, YU Jiandong, CAIRNS T C, et al. Comprehensive improvement of sample preparation methodologies facilitates dynamic metabolomics of Aspergillus niger [J/OL]. Biotechnology Journal, 2018, 14(3): 1800315. DOI: 10.1002/biot.201800315.
[45]ZABEK A, JUNKA A, SZYMCZYK P, et al. Metabolomics analysis of fungal biofilm development and of arachidonic acid-based quorum sensing mechanism [J]. Journal of Basic Microbiology, 2017, 57(5): 428-439.
[46]LOWE R G T, ALLWOOD J W, GALSTER A M, et al. A combined 1h nuclear magnetic resonance and electrospray ionization-mass spectrometry analysis to understand the basal metabolism of plant-pathogenic Fusarium spp. [J]. Molecular Plant Microbe Interact, 2010, 23(12): 1605-1618.
[47]CHRISTOPH J B, PATRICK K, FABIEN L, et al. Sampling for metabolome analysis of microorganisms [J]. Analytical Chemistry, 2007, 79(10): 3843-3849.
[48]TOMAS C, MARTA V, ZBYNEK D, et al. Rapid LC-MS-based metabolomics method to study the Fusarium infection of barley [J]. Journal of Separation Science, 2014, 37(8): 912-919.
[49]SHEN Yang, FATEMEH T, TANG Leihan, et al. Quantitative metabolic network profiling of Escherichia coli: an overview of analytical methods for measurement of intracellular metabolites [J]. Trends in Analytical Chemistry, 2016, 75: 141-150.
[50]PHUONG A N, CAROLINE S, MARIE L, et al. Study of in vitro interaction between Fusarium verticillioides and Streptomyces sp. using metabolomics [J]. Folia Microbiologica, 2020, 65(2): 303-314.
[51]LIU Caixiang, CHEN Fangfang, ZHANG Jingtao, et al. Metabolic changes of Fusarium graminearum induced by TPS gene deletion [J]. Journal of Proteome Research, 2019, 18(9): 3317-3327.
[52]KARPE A V, DUNN M S, TAYLOR M C T. et al. Nitrogen deprivation in Fusarium oxysporum promotes mycotoxin production via intermediates in the Krebs cycle and unreported methylmalonyl-CoA mutase activity [J/OL]. Metabolomics, 2018, 14(12): 160. DOI: 10.1007/s11306-018-1459-0.
[53]趙春霞, 许国旺. 基于液相色谱-质谱技术的代谢组学分析方法新进展[J]. 分析科学学报, 2014, 30(5): 761-766.
[54]CHEN Wei, GONG Liang, GUO Zilong, et al. A novel integrated method for large-scale detection, identification, and quantification of widely targeted metabolites: application in the study of rice metabolomics [J]. Molecular Plant, 2013, 6(6): 1769-1780.
[55]WOLFENDER J L, MARTI G, THOMAS A, et al. Current approaches and challenges for the metabolite profiling of complex natural extracts [J]. Journal of Chromatography A, 2015, 1382: 136-164.
[56]HAUTBERGUE T, JAMIN E L, DEBRAUWER L, et al. From genomics to metabolomics, moving toward an integrated strategy for the discovery of fungal secondary metabolites [J]. Natural Product Reports, 2018, 35(2): 147-173.
[57]SEVASTOSA A, KALAMPOKISA I F, PANAGIOTOPOULOUB A, et al. Implication of Fusarium graminearum primary metabolism in its resistance to benzimidazole fungicides as revealed by 1H NMR metabolomics [J]. Pesticide Biochemistry and Physiology, 2018, 148: 50-61.
[58]ISSAQ H J, VAN Q N, WAYBRIGHT T J, et al. Analytical and statistical approaches to metabolomics research [J]. Journal of Separation Science, 2015, 32(13): 2183-2199.
[59]RODRIGUES A M, MIGUE C, CHAVES I, et al. Mass spectrometry‐based forest tree metabolomics [J]. Mass Spectrometry Reviews, 2019, 47(2): 126-157.
[60]ZHAO Liang, THOMAS H. Metabonomics and toxicology [J]. Methods in Molecular Biology, 2015, 1277: 209-231.
[61]LIU Xinyu, ZHOU Lina, SHI Xianzhe, et al. New advances in analytical methods for mass spectrometry-based large-scale metabolomics study [J/OL]. Trends in Analytical Chemistry, 2019, 121: 115665. DOI: 10.1016/j.trac.2019.115665.
[62]CAJKA T, FIEHN O. Increasing lipidomic coverage by selecting optimal mobile-phase modifiers in LC-MS of blood plasma [J/OL]. Metabolomics, 2016, 12(2): 34. DOI: DOI 10.1007/s11306-015-0929-x.
[63]LI Yong, RUAN Qiang, LI Yanli, et al. A novel approach to transforming a non-targeted metabolic profiling method to a pseudo-targeted method using the retention time locking gas chromatography/mass spectrometry-selected ions monitoring [J]. Journal of Chromatography A, 2012, 1255: 228-236.
[64]TSUGAWA H, TSUJIMOTO Y, SUGITATE K, et al. Highly sensitive and selective analysis of widely targeted metabolomics using gas chromatography/triple-quadrupole mass spectrometry [J]. Journal of Bioscience and Bioengineering, 2014, 117(1): 122-128.
[65]HALKET J M, WATERMAN D, PRZYBOROWSKA A M, et al. Chemical derivatization and mass spectral libraries in metabolic profiling by GC/MS and LC/MS/MS [J/OL]. Journal of Experimental Botany, 2005, 56(410): 219. DOI: 10.1093/jxb/eri069.
[66]WEN Jing, YANG Lina, QIN Feng, et al. An integrative UHPLC-MS/MS untargeted metabonomics combined with quantitative analysis of the therapeutic mechanism of Si-Ni-San [J]. Analytical Biochemistry, 2019, 567: 128-135.
[67]MARINA G, BASEM K, FRANK S, et al. Comprehensive analysis of the Alternaria mycobolome using mass spectrometry-based metabolomics [J/OL]. Molecular Nutrition Food Research, 2020, 64(3): 1900558. DOI: 10.1002/mnfr.201900558.
[68]ZHOU Juntuo, LIU Huiying, LIU Yang, et al. Development and evaluation of a parallel reaction monitoring strategy for large-scale targeted metabolomics quantification [J]. Analytical Chemistry, 2016, 88(8): 4478-4486.
[69]FERNAND M G, ROULLIER C, GUITTON Y, et al. Fungi isolated from Madagascar shrimps-investigation of the Aspergillus niger metabolism by combined LC-MS and NMR metabolomics studies [J]. Aquaculture, 2017, 479: 750-758.
[70]GOODACRE R. Metabolomics of a superorganism [J]. Journal of Nutrition, 2007, 137: 259S-266S.
[71]LIU Xiaojing, LOCASALE J W. Metabolomics: A primer [J]. Trends in Biochemical Sciences, 2017, 42(4): 274-284.
[72]KOUSKOUMVEKAKI I, YANG Zhiyong, JNSDTTIR S O, et al. Identification of biomarkers for genotyping Aspergilli using non-linear methods for clustering and classification [J/OL]. BMC Bioinformatics, 2008, 9: 59. DOI: 10.1186/1471-2105-9-59.
[73]MASTRANGELO A, FERRARINI A, REY S F, et al. From sample treatment to biomarker discovery: A tutorial for untargeted metabolomics based on GC-(EI)-Q-MS [J]. Analytica Chimica Acta, 2015, 900: 21-35.
[74]TAYLOR J, KINGR D, ALTMANN T, et al. Application of metabolomics to plant genotype discrimination using statistics and machine learning [J]. Bioinformatics, 2002, 18(S2): S241-S248.
[75]SEVASTO A, KALAMPOKIS I F, PANAGIOTOPOULOU A, et al. Fusarium graminearum 1H NMR metabolomics [J]. Data in Brief, 2018, 19: 1162-1165.
[76]HUANG Baoming, ZHA Qinglin, CHEN Tingbo, et al. Discovery of markers for discriminating the age of cultivated ginseng by using UHPLC-QTOF/MS coupled with OPLS-DA [J]. Phytomedicine, 2018, 45: 8-17.
[77]俞邱豪, 张九凯, 叶兴乾, 等. 基于代谢组学的食品真实属性鉴别研究进展[J]. 色谱, 2016, 34(7): 657-664.
[78]KALIVODOV A, HRON K, FILZMOSER P, et al. PLS-DA for compositional data with application to metabolomics [J]. Journal of Chemometrics, 2015, 29(1): 21-28.
[79]王清利. 基于UHPLC-MS/MS技术的细胞代谢组学方法的建立及应用[D]. 郑州:郑州大学, 2018.
[80]GU Qiongnan, YUAN Qinfeng, ZHAO Dian, et al. Acetyl-coenzyme A synthetase gene ChAcs1 is essential in lipid metabolism, carbon utilization and virulence of the hemibiotrophic fungus Colletotrichum higginsianum [J]. Molecular Plant Pathology, 2019, 20(1): 107-123.
[81]CAO Huijuan, HUANG Pengyun, YAN Yuxin, et al. The basic helix-loop-helix transcription factor Crf1 is required for development and pathogenicity of the rice blast fungus by regulating carbohydrate and lipid metabolism [J]. Environmental Microbiology, 2018, 20(9): 3427-3441.
[82]BRISSON V L, ZHUANG Weiqin, ALVAREZ-COHEN L. Metabolomic analysis reveals contributions of citric and citramalic acids to rare earth bioleaching by a Paecilomyces fungus [J/OL]. Frontiers in Microbiology, 2020, 10: 3008. DOI: 10.3389/fmicb.2019.03008.
[83]AGUILAR-PONTES M V, BRAND J, MCDONNELL E, et al. The gold-standard genome of Aspergillus niger NRRL 3 enables a detailed view of the diversity of sugar catabolism in fungi [J]. Studies in Mycology, 2018, 91: 61-78.
(責任编辑:杨明丽)
