选择性SGLT2抑制剂的研究进展
2021-10-09郭家钰巫振坤贺岩吴成军孙铁民沈阳药科大学基于靶点的药物设计与研究教育部重点实验室制药工程学院沈阳006北部战区总医院药剂科沈阳0840
郭家钰,巫振坤,贺岩,吴成军*,孙铁民*(.沈阳药科大学基于靶点的药物设计与研究教育部重点实验室制药工程学院,沈阳 006;.北部战区总医院药剂科,沈阳 0840)
糖尿病(diabetes mellitus,DM)是一种与代谢功能障碍相关的常见疾病,通常发生在胰腺无法产生足够的胰岛素或胰岛素因抵抗而无法正常发挥降血糖作用的患者中[1]。截止至2020年,全球糖尿病患者约4.25 亿,预计到2030年,糖尿病患者数量将增加到约5.52 亿。糖尿病患病人数的增长与人们生活条件的改变息息相关。糖尿病的发生通常伴随着肥胖、高血压、心血管疾病或动脉粥样硬化等各种其他慢性疾病和病症,从而导致患者生活质量下降[2-4]。
美国糖尿病协会(ADA)和世界卫生组织将糖尿病分为4 个类别:1 型糖尿病(T1DM)、2型糖尿病(T2DM)、妊娠糖尿病以及其他特定类型的糖尿病。T1DM 是由T 细胞介导的胰岛β细胞自身免疫破坏引起的胰岛素缺乏所导致的,多发于青少年。根据ADA 的相关统计,T1DM 仅占糖尿病患者的5%~10%。T2DM 是由胰岛素分泌的减退、胰岛素抵抗(IR)或两种情况同时发生导致的碳水化合物、脂质和蛋白质代谢失调所引起的。在所有糖尿病患者中,T2DM 占比超过90%[5]。尽管T2DM 的发病机制尚不明确,但遗传因素、生活环境、肥胖、高血压在一定程度上与T2DM 的发生相关[6]。
研究表明,通过改变生活方式和控制饮食的方法可以辅助调控患者的血糖,但是这种方式对血糖水平的调控是有限的。并且,随着糖尿病的发展,仅仅依靠传统药物治疗并不能取得理想的效果。在1950 到1990年代中期,接近40年的时间里,只有胰岛素、磺脲类药物和双胍类药物用于糖尿病治疗。在1995年之后,一些新型的糖尿病药物才逐步被开发并应用于临床[7-9](见图1),本文将对SGLT2 及其作用机制进行综述,并与其他降糖药物进行比较,为新型选择性SGLT2抑制剂的研发提供参考。
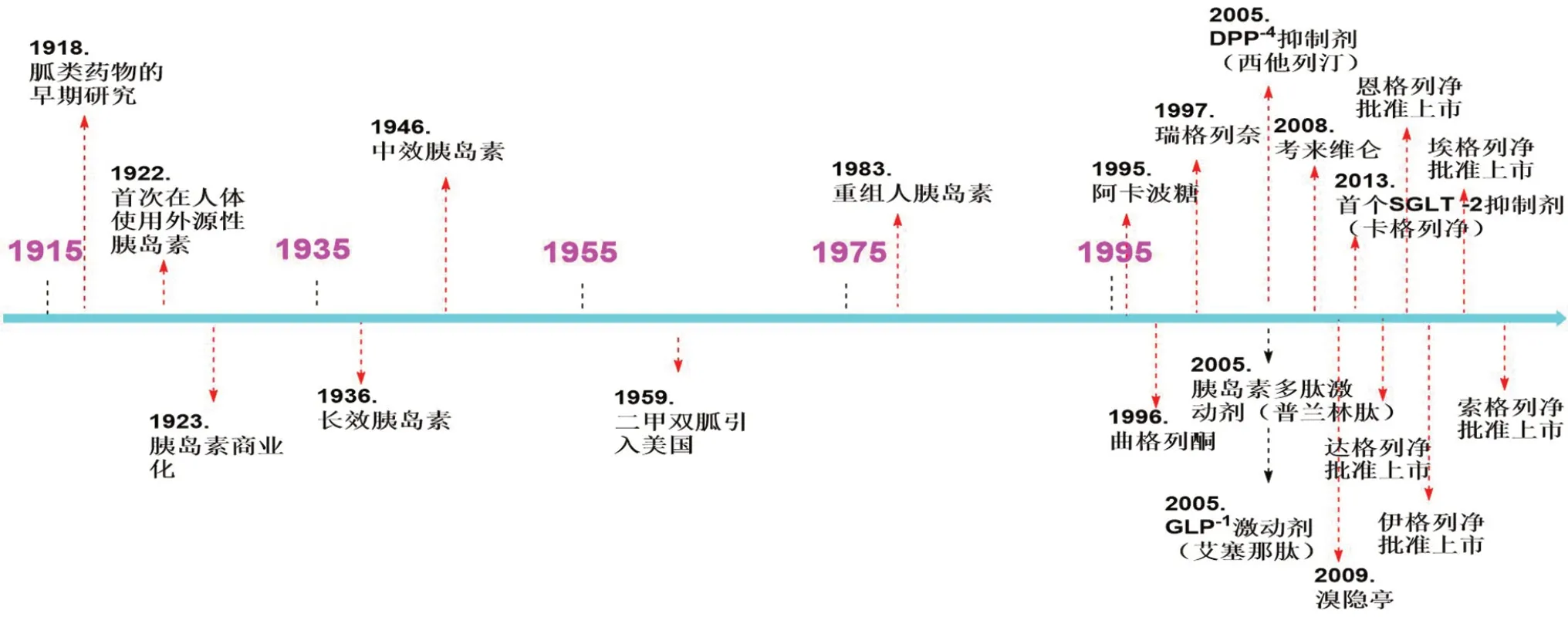
图1 近年来降血糖药物的发展[9]Fig 1 Development of hypoglycemic drugs in recent years[9]
1 传统降血糖药物
1.1 促进胰岛素合成、分泌类药物
1.1.1 磺酰脲类药物 临床采用此种药物治疗T2DM 已经近半个世纪。磺酰脲类药物主要通过刺激胰岛β细胞,促进其释放胰岛素,但不会影响患者自身胰岛素的合成。长期服用此药,会引起一定的低血糖反应,且此类药物仅局限于胰岛β细胞功能尚存的患者,对胰岛β细胞功能受损的患者没有疗效[10]。
1.1.2 胰高血糖素样肽1(GLP1)受体激动剂与葡萄糖依赖性促胰岛素多肽类似,可促进肠促胰岛素激素释放,促进胰岛素的分泌从而降低血糖。其对胰岛β细胞功能受损的患者同样无法产生疗效[11]。
1.2 胰岛素增敏剂
1.2.1 双胍类药物 此类药物通过减少肝糖分解,降低糖异生,增加患者体内胰岛素的敏感性来发挥作用,并且不会造成胰岛功能减退。同时,双胍类药物还能有效降低胰岛素抵抗作用,并显著减轻T2DM 患者的体重[12]。
1.2.2 噻唑烷二酮类药物 此类药物能够有效作用于患者肝脏系统,激活胰岛素抵抗、脂肪组织,进一步增加胰岛素自身的敏感性,降低患者血糖水平。临床研究表明,噻唑烷二酮类药物具有降低胰岛素抵抗的作用。但是,在部分患者中发现,噻唑烷二酮类药物会引起液体潴留,继而发展为充血性心力衰竭(congestive heart failure,CHF)[13]。
1.3 α-葡萄糖苷酶抑制剂
α-葡萄糖苷酶抑制剂可通过竞争性抑制位于小肠中的α-葡萄糖苷酶,延缓碳水化合物的吸收。α-葡萄糖苷酶抑制剂仅改变餐后葡萄糖水平,不改变空腹血浆葡萄糖水平,所以导致该药物调控糖化血红蛋白(HbA1c)的能力下降。此类药物的已知不良反应包括胃部不适,例如腹泻、腹胀和腹部绞痛等[14]。
1.4 钠/葡萄糖共转运蛋白(SGLT)抑制剂
SGLT 属于膜蛋白家族(见表1),可促进葡萄糖、氨基酸和维生素等物质的转运,促进各种离子在近曲小管和肠上皮的吸收。SGLTs 转运的葡萄糖中有大约90%会在肾脏中被重新吸收,因此通过抑制这些转运蛋白可以阻止葡萄糖的重吸收,从而导致糖尿增加,血糖降低,由于在这一过程中不涉及胰岛素,所以该抑制剂可以在T2DM 的任何阶段使用。该机制是改善糖尿病患者血糖控制最有前途的治疗方法之一[15]。
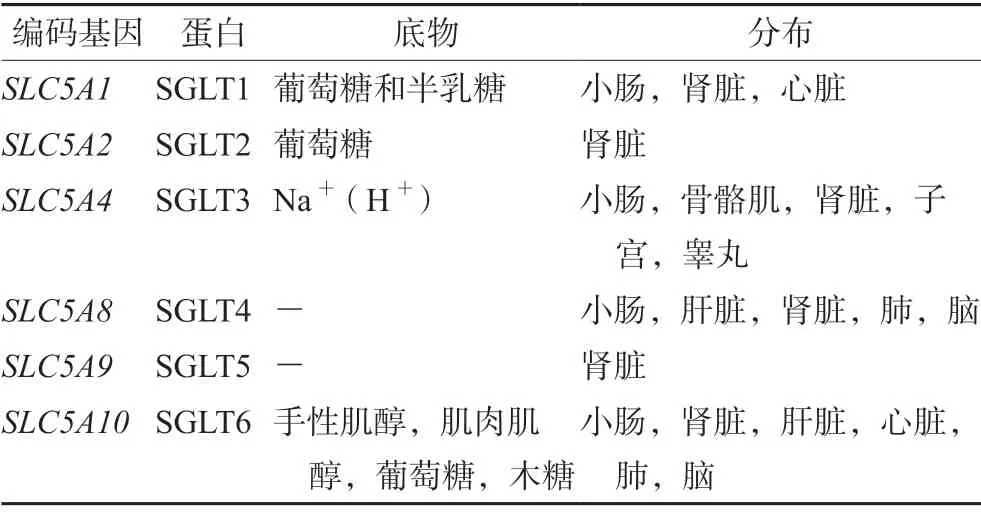
表1 SGLTs 蛋白在体内分布Tab 1 Distribution of SGLTs proteins in the body
SGLT2 主要分布在肾脏近曲小管S1 部位,负责肾脏中约90%葡萄糖的重吸收,抑制SGLT2 可以促进糖尿病患者尿糖的排出,因此SGLT2 抑制剂被认为是一种新型的具有独特作用机制的抗糖尿病药物。SGLT2 抑制剂也被发现在胰岛素抵抗和胰岛β细胞细胞功能障碍病例中具有很好的疗效,为糖尿病治疗提供了一种全新的治疗选择[16]。
2 SGLT2 抑制剂研究进展
2.1 SGLT2 天然活性产物
根皮苷(phlorizin)是由法国的研究人员于19世纪50年代在苹果树皮中得到的一种查尔酮类天然产物,并一度被认为是SGLT1 和SGLT2 的非特异性抑制剂。根皮苷能够减少尿液中葡萄糖的重吸收并且增加肾脏对葡萄糖的清除,从而降低了高血糖和糖中毒的风险。但是,根皮苷会非选择性地抑制SGLT1,且由于其不良反应和口服生物利用度的问题,所以它并没有被开发成抗糖尿病药物[17-19]。此外,研究发现根皮苷的水解代谢产物根皮素可以阻断葡萄糖转运通道1(glucose transporter 1,GLUT1),严重干扰了葡萄糖在其他组织器官中的摄取和利用,从而影响患者的生理健康[20]。
在20世纪中叶之前,根皮苷被一度认为是造成肾脏和小肠中红细胞停止吸收葡萄糖的罪魁祸首。但是,在20世纪末期,随着对SGLT2 研究的深入,阐明了根皮苷对于糖尿和肾脏的作用机制[16,21],于1990年在根皮苷的基础上发现了T-1095,T-1095 在体内代谢成活性成分T-1095A 发挥作用。在活性测试中发现T-1095A 的抑制活性(SGLT1:IC50=0.20 µmol·L-1,SGLT2:IC50=0.05µmol·L-1)与根皮苷的抑制活性(SGLT1:IC50=0.16 µmol·L-1,SGLT2:IC50=0.16 µmol·L-1)接近,但是仍然没有克服O-葡萄糖苷类似物药代动力学不稳定的问题,故止步于此[22](见图2)。

图2 天然产物根皮苷及T-1025 的化学结构Fig 2 Chemical structure of natural product phlorizin and T-1025
2.2 已上市的SGLT2 抑制剂(见图3)

图3 上市SGLT2 抑制剂的化学结构Fig 3 Chemical structure of commercially available SGLT2 inhibitors
2.2.1 卡格列净(canagliflozin) 第一个经FDA 批准上市的SGLT2 抑制剂,其与二甲双胍的复方制剂(商品名:Invokamet)在2015年即实现了13.08亿元的巨额销售;卡格列净通过抑制肾近曲小管的葡萄糖重吸收使血糖降低[23]。研究显示,与安慰剂组相比,每日分别给予100 和300 mg 的卡格列净能明显降低HbA1c 水平;当卡格列净单独使用时,HbA1c 水平分别下降了0.91%和1.16%;在与磺酰脲类、二甲双胍、二甲双胍加吡格列酮、二甲双胍加磺酰脲类和胰岛素联合使用时,HbA1c下降了0.62%~0.925%(排除特殊人群,例如老年人和肾功能不全者)[24]。与对照组比较,大量给予100 和300 mg 卡格列净治疗的患者体重减轻了0.4%~3.3%,收缩压(SBP)降低0.1~7.9 mmHg,高密度脂蛋白胆固醇(HDL-C)水平升高。
在口服卡格列净或其他SGLT2 抑制剂时会产生一些不良反应,如口渴,尿量增加,低密度脂蛋白(LDL)胆固醇水平升高以及高血压和泌尿生殖道感染的发生率增加等。因此,在患有肾功能不全,肾小球滤过率(GFR)降低的人群中禁用卡格列净[25]。
FDA 在2015年5月发出警告,卡格列净和某些SGLT2 抑制剂可能会引起酮症酸中毒。2015年9月,FDA 声明卡格列净存在着使骨密度降低从而导致骨折的风险[26]。
2.2.2 恩格列净(empagliflozin) 是由勃林格殷格翰和礼来公司共同开发的一种SGLT2 抑制剂,该药物于2014年被FDA 批准上市。恩格列净具有与根皮苷相似的C-葡萄糖苷结构,对SGLT2有更高的选择性(是SGLT1 的2500 多倍)[27]。
相比于安慰剂对照组,恩格列净组患者的SBP 和体重都明显下降。相比于单一使用恩格列净,联合使用25 mg 的恩格列净和二甲双胍时体重和SBP 明显降低。后续的临床研究表明,恩格列净可以降低中风、心肌梗死和心血管死亡的风险,这使恩格列净成为第一个FDA 批准的用于降低心血管死亡风险的SGLT2 抑制剂[28]。
2.2.3 达格列净(dapagliflozin) 是由百时美施贵宝和阿斯利康公司共同研发的C-芳基糖苷类SGTL-2 抑制剂,在2014年1月8日,FDA 批准了达格列净与饮食和运动一起用于控制糖尿病患者的血糖的治疗方案。2014年10月,FDA 又批准了达格列净与盐酸二甲双胍联合治疗的方案。
达格列净的常见不良反应包括重度糖尿症、疲倦且体重减轻伴随着脱水以及糖尿病酮症酸中毒(DKA)的风险增加。除此之外,达格列净还会因为增加尿液中的含糖量而加剧糖尿病所引起的感染[29]。2017年3月10日,达格列净在中国上市,成为首个在国内上市的SGLT2 抑制剂。
2.2.4 伊格列净(ipragliflozin) 是2014年1月在日本获得批准上市的口服SGLT2 抑制剂。伊格列净既可单独用于T2DM 的治疗,也可与其他降血糖药物(例如二甲双胍、磺酰脲类或α-葡萄糖苷酶抑制剂和吡格列酮)联合使用。FDA 批准上市了25 和50 mg 两种规格的伊格列净片,如果50 mg 的伊格列净疗效不足,则伊格列净剂量可以增加到每日100 mg[30]。2018年12月,安斯泰来制药公司和日本Kotobuki 制药公司在日本获得了伊格列净L-脯氨酸用于治疗T1DM 的批准。体外药理研究发现,伊格列净可以竞争性地抑制SGLT2,并且对SGLT2 的选择性比SGLT1 的选择性高254 倍[31]。
2.2.5 托格列净(tofogliflozin) 是由日本中外制药开发的一种用于治疗T2DM 的口服SGLT2抑制剂[32],其既可以作为降糖药物单独使用也可以与其他药物联合使用治疗糖尿病[33]。现已批准20 mg 剂量的托格列净用于治疗T2DM[34]。
2.2.6 鲁格列净(luseogliflozin) 是由日本开发的选择性SGLT2 抑制剂。鲁格列净的批准剂量有2.5 和5 mg 两种,一般建议每日起始剂量为2.5 mg,如果效果不佳,则增加剂量到每日5 mg。在长期治疗中,无论是作为单一疗法还是与其他口服抗高血糖药物联合使用,鲁格列净均表现出较好的治疗效果[35]。
2.2.7 埃格列净(ertugliflozin) 是默克公司和辉瑞公司联合开发的新型口服SGLT2 抑制剂。使用5 mg 和15 mg 剂量的埃格列净治疗后,HbA1C 分别降低了0.69%和0.76%(相比于安慰剂对照组)。研究还发现,相比于单独使用埃格列净或西他列汀,二者联合使用可使HbA1C降至7.0%以下。2017年,埃格列净(商品名Steglatro)被FDA 批准上市[36-37]。
2.2.8 索格列净 2016年Lexicon 制药公司在Ⅲ期临床实验中发现,索格列净可显著降低T1DM 患者HbA1C 的水平,治疗24 周后,每日200、400 mg 剂量的索格列净可使HbA1C 相比于基线分别降低0.43%、0.49%。而安慰剂只能使HbA1C 降低0.08%。索格列净是同类产品中第一个同时抑制SGLT1 和SGLT2 的双靶点抑制剂[38-39]。2019年4月26日,索格列净获得欧洲药物管理局批准上市,成为第一个获批上市的SGLT1/SGLT2 双靶点抑制剂。
近年研究发现,现有的SGLT2 抑制剂除了能降血糖,还能降低心力衰竭的发生风险,减轻体重和保护肾脏[40],这使得SGLT2 抑制剂成为了具有开发潜力的降血糖药物,在糖尿病药物市场中占据了越来越大的份额。
2.3 处于研发阶段的 SGLT2 抑制剂
2.3.1 三唑类(见图4) 1,2,3-取代和1,2,4-取代的三唑类化合物是很多药物(例如抗病毒剂、抗过敏剂、抗HIV 和抗菌剂)的核心结构。
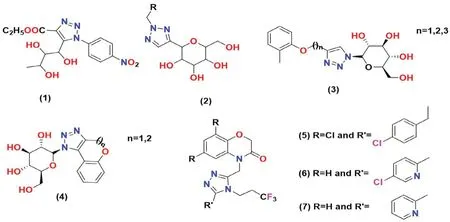
图4 三唑类SGLT2 抑制剂化学结构Fig 4 Chemical structure of triazole SGLT2 inhibitors
Putapatri 等[41]设计并合成了葡萄糖苷类衍生物,将L-鼠李糖的C-核苷与1,2,3-三唑核偶联。使用葡萄糖摄取测定法(评估SGLT1 和SGLT2抑制效果)对这些衍生物进行降血糖活性筛选,结果发现对硝基苯基衍生物1 对SGLT2 具有抑制活性,其IC50值为125.9 nmol·L-1。
Li 等[42]使用“点击化学”的策略,设计并合成一系列三唑类葡萄糖苷衍生物并评估了其对糖尿病的影响,发现了含有三唑糖苷配基核心的C-葡萄糖苷的化合物2具有良好的SGLT2抑制活性,且几乎所有合成的三唑C-葡萄糖苷均会增加尿葡萄糖排泄(urinary glucose excretion,UGE),但是尿量的增加都小于达格列净对照组。
Bai 等[43]首次合成了一种新型的三唑-N-糖苷衍生物,并测试了其对葡萄糖转运的抑制作用(以达格列净作对照)。实验结果表明,将三唑部分与N-葡萄糖苷或N-半乳糖苷3 和4 相连的化合物显示出了很强的抑制活性。
Du 等[44]合成了一种新的非根皮苷类的SGLT2抑制剂,并进行了SGLT 的相关活性测试。在对人的SGLT2 进行筛选时,发现化合物5 是最有效的抑制剂(IC50=10 nmol·L-1)。化合物5 还对SGLT1 表现出很好的选择性(IC50=9 μmol·L-1),当在大鼠体内进行实验时,观察到了较低的微粒体稳定性和较高的清除率。
该系列的SAR 分析表明,含有苯并恶嗪酮的化合物与芳环相连的结构显示SGLT2 抑制剂有效;通过在苯并恶嗪酮的C-8 位上连接一些极性基团(如氰基或吡啶基团)合成的化合物6(IC50=3 nmol·L-1;溶解度=1.1 μg·mL-1)和化合物7(IC50=12 nmol·L-1;溶解度=64 μg·mL-1)与先导化合物5 相比,均表现出更好的抑制活性[44]。
2.3.2 吲哚类(见图5) 使用葡萄糖取代D-木糖,并在第三位引入吲哚基团,得到的N-吲哚木糖苷类化合物,表现出了很强的SGLT2 抑制剂活性。此外,C-吲哚葡萄糖苷衍生物也被发现是良好的SGLT2 抑制剂[45]。

图5 吲哚类SGLT2 抑制剂化学结构Fig 5 Chemical structure of indole SGLT2 inhibitors
Zhang 等[45]通过修饰先前报道的降血糖混合物8 合成了一系列吲哚-O-和C-葡萄糖苷化合物。通过对新化合物进行体外SGLT1 和SGLT2的抑制活性测试,筛选出了化合物9 和10,分别是有效的SGLT2 抑制剂和SGLT1/SGLT2 双靶点抑制剂,其具有促进UGE 的功效。进一步测试了这两种化合物在ZDF 雄性大鼠中的疗效,发现这两种新抑制剂的药代动力学参数与化合物8 相似,但药效却显著降低。SAR 分析表明,将化合物9 中与2,3-二氢苯并呋喃部分的连接部分缩短得到的化合物11 对SGLT2 抑制活性减弱,甚至消失;而当2,3-二氢苯并呋喃部分被另一个芳香基团修饰后,得到的化合物展现出较强的SGLT2 抑制活性。该研究还表明,异头氧(糖苷氧)的存在对于SGLT 的抑制活性至关重要,将其取代所得到的C-葡萄糖苷类化合物的活性显著降低。
Chu 等[46]合成了一系列N-吲哚基葡萄糖苷化合物,在以根皮苷为阳性对照的实验中测试了SGLT2 的体外抑制活性。SAR 分析表明,通过修饰C6-葡萄糖得到的化合物抗糖尿病活性明显升高。在筛选中发现C6位被酰胺修饰的化合物12 具有优良的SGLT2(化合物12,EC50=42 nmol·L-1;根皮苷EC50=123 nmol·L-1)抑制活性,但对SGLT1(化合物12,EC50=1412 nmol·L-1;根皮苷,EC50=153 nmol·L-1)的选择性较差。
Li 等[47]使用全新的方法合成了N-糖苷吲哚类衍生物13。在化合物13 的基础上使用F-C 反应进一步修饰得到了化合物14。经测试,这两个化合物均表现出了优良的SGLT2 抑制活性。
Yao 等[48]设计并合成了一系列吲哚基木糖苷衍生物。在实验中观察到含有C-糖苷键的化合物不能被胃肠道葡萄糖苷酶水解,从而避免了胃肠道(gastrointestinal tract,GIT)对这一类化合物的清除,保持着较好的抑制活性。在研发新的SGLT抑制剂时,采用D-木糖取代C-葡萄糖苷中的葡萄糖合成了一系列的吲哚木糖衍生物,结果发现化合物15 是最有效的SGLT2 抑制剂[SGLT2:EC50=(47±3)nmol·L-1,SGLT1:EC50=(282±11)nmol·L-1];化合物16 是最有效的SGLT1 抑制剂[SGLT2:EC50=(50±11)nmol·L-1,SGLT1:EC50=(55±5)nmol·L-1]。
SAR 研究表明,在环丙基苯基的对位连着吲哚的N时,对吲哚基团的7 位进行修饰得到的化合物15 和16 表现出最好的抑制活性。体内研究表明,化合物15 代谢稳定,清除率低,具有良好的降血糖活性(与对照药物相比,化合物15 使糖尿病大鼠血糖水平降低35%以上)[48]。
2.3.3 噻吩类(见图6) Lee 等[49]设计并合成了两种不同类型的含噻吩环的C-芳基葡萄糖苷衍生物,并对其作为SGLT2 抑制剂进行了活性评价。在这些衍生物中,发现化合物17 是最有效的抑制剂,其IC50值为4.47 nmol·L-1。这一结果表明,当苄基第4 位与烷基链相连时(具有适当数量的碳原子)得到的化合物表现出良好的体外SGLT2 抑制活性。

图6 噻吩类SGLT2 抑制剂化学结构Fig 6 Chemical structure of thiophene SGLT2 inhibitors
Koga 等[50]通过将卡格列净中的苯环替换为不同的部分,例如吡啶、嘧啶和五元噻吩偶联杂芳烃,得到了一系列衍生物。在实验中发现化合物18 活性最好,其对SGLT2(IC50=2 nmol·L-1)表现出很强的抑制活性(卡格列净IC50=2.2 nmol·L-1)。
2.3.4 哒嗪类(见图7) Min 等[51]设计并合成了具有哒嗪环的葡萄糖苷芳香族衍生物。在活性测试中发现化合物19 和20 具有明显的SGLT2 抑制活性,IC50值分别为11.4 和13.4 nmol·L-1。此外,含有甲硫基的化合物20 在正常SD 大鼠体内表现出显著的SGLT2 抑制活性。
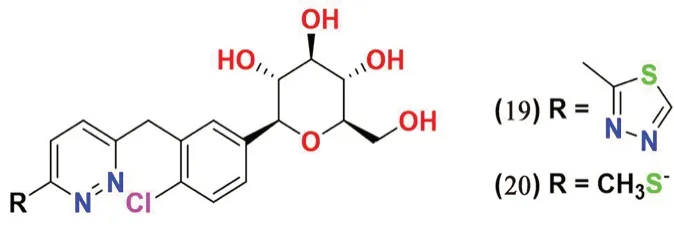
图7 哒嗪类SGLT2 抑制剂化学结构Fig 7 Chemical structure of pyridazine SGLT2 inhibitors
2.3.5 噻唑和噻二唑类(见图8) Lee 等[52]合成了一种含有呋喃基团的噻唑类化合物,该系列化合物在体外的活性测试中表现出了纳摩尔级别的SGLT2 抑制活性。筛选发现化合物21 和22是最有效的SGLT2 抑制剂,IC50值分别为0.934 nmol·L-1和0.797 nmol·L-1。

图8 噻唑和噻二唑类SGLT2 抑制剂化学结构Fig 8 Chemical structure of thiazole and thiadiazole SGLT2 inhibitors
Kang 等[53]合成了一系列将噻唑环与C-葡萄糖苷相连的噻唑类化合物,并且对噻唑的C-4 或C-5 位进行了卤素修饰。体外SGLT2 抑制实验发现,苄基噻唑衍生物23 具有显著的SGLT2 抑制剂活性(IC50值为121 nmol·L-1)。
除此之外,研究人员还设计并合成了一系列1,3,4-噻二唑偶联的C-芳基葡萄糖苷类化合物,并对其进行了SGLT2 活性测试。其中化合物24~26 对SGLT2 的体外抑制活性较好,IC50值分别为4.06、7.03 和3.51 nmol·L-1[54]。
SAR 分析表明,用1,3,4-噁二唑取代达格列净末端的苯环得到的化合物降血糖活性消失,而1,3,4-噻二唑环取代则增加了抗糖尿病活性。此外,噻二唑环上的线性脂肪链的取代使抗糖尿病活性下降,采用更大的基团取代时则导致抗糖尿病活性完全丧失。如果在苯环的C-4'位置引入类似卤素的亲脂性小取代基,则会使抗糖尿病活性提高。经过筛选得到的活性较好的化合物(如含有吡嗪环的化合物24、含有呋喃环的化合物25 和含有噻吩环的化合物26)的共有结构是杂环,提示这种杂环对SGLT2 的抑制活性很重要[54]。
2.3.6 螺环类(见图9) Lv 等[55]在C-芳基葡萄糖苷的基础上,在邻位引入螺环合成了一系列新的化合物,并对该系列化合物进行了体外SGLT1和SGLT2 活性测试。化合物27 和28 表现出了显著的体外活性,对SGLT2 的IC50值分别为0.3 和3.8 nmol·L-1,对SGLT1的IC50值分别为3.1和0.7 nmol·L-1,化合物27 对SGLT2 选择性更高,而化合物28 对SGLT1 表现出更好的选择性。测试结果显示,当B 环的C-4 位置被氯取代的螺环修饰时表现出更好的抗糖尿病活性。

图9 螺环类SGLT2 抑制剂化学结构Fig 9 Chemical structure of spirocyclic SGLT2 inhibitors
Xu 等[56]合成了一系列的O-螺环-C-芳基葡萄糖苷化合物,其中螺环基团连接到C-芳基葡萄糖苷的2'和6'位置。以1 μmol·L-1为单一剂量测试了体外对SGLT1 和SGLT2 活性,6'-O-螺环取代的C-芳基糖苷衍生物对SGLT2 抑制活性更好;2'-O-螺环衍生物对此靶标没有显示出明显活性。两种6'-O-螺环衍生物化合物29 和30 被筛选为有效的SGLT2 抑制剂,IC50值分别为71 和6.6 nmol·L-1。SAR 表明,苯环上4'位的取代基对SGLT2 抑制活性至关重要,4'位没有取代的化合物29 相比于4'位氯取代的化合物30 表现出10 倍以上的活性丧失。
Robinson 等[57]制备了一系列C-5 螺环衍生物,通过评估其体外SGLT2 抑制活性,筛选出化合物31~35,其中化合物33 和35 在大鼠体内显示出良好的功效。
Mascitti 等[58]合成了在吡喃糖环第5 位被螺环修饰的C-糖苷衍生物。通过评估这些衍生物对SGLT1 和SGLT2 的抑制活性和选择性,得到了SGLT2 抑制活性较好的化合物36~37,IC50值分别为(6.6±2.5)、14 和(3.4±0.8)nmol·L-1。
2.3.7 苯基类(见图10) Xu 等[59]合成了一系列在第二个碳原子上具有取代基的C-芳基葡萄糖苷衍生物,这些衍生物在体外细胞测试中展现对SGLT1 和SGLT2 较好抑制活性。其中化合物38 和39 活性较好,对SGLT1 的IC50分别为0.9、1.2 nmol·L-1,对SGLT2 的IC50分别为2、4.2 nmol·L-1。化合物38 在体外实验中表现出7 倍于达格列净的SGLT2 的抑制活性和11 倍选择性。

图10 苯基类SGLT2 抑制剂化学结构Fig 10 Chemical structure of phenyl SGLT2 inhibitors
SAR 表明,与糖苷键相邻的苯环的第2 个碳原子(邻位)上引入取代基不仅会增加SGLT2 的抑制活性,而且还会提高化合物对SGLT2 的选择性[59]。
Min 等[60]设计并且合成了一类引入了大环结构的C-芳基葡萄糖苷类SGLT2 抑制剂。化合物40、41 在体外实验中对SGLT2 的抑制IC50值为0.778 nmol·L-1和0.899 nmol·L-1[达格列净IC50=(1.35±0.15)nmol·L-1],但在SD 大鼠体内中仅表现出中等活性。
Li 课题组[61]合成了一系列具有二氧杂双环的C-芳基葡糖苷衍生物,在体外测试了其对SGLT2 的抑制活性并筛选出了化合物42,IC50值为700 nmol·L-1。
2.3.8 薁类(见图11) Ikegai 等[62]设计并合成了一系列将薁环连接到与糖部分直接相连的苯环上的化合物并且在体外评估其抗SGLT1和SGLT2 活性。其中化合物43 和44 表现出对SGLT2 良好的选择性和抑制活性(IC50=22 nmol·L-1和8.9 nmol·L-1),化合物44 还在糖尿病大鼠和小鼠的体内测试中表现出较好的活性。SAR 分析表明,邻位的羟基取代化合物(化合物44)展现出对于SGLT2 的更好的抑制活性。化合物44 的单胆碱化合物45 正在进行进一步的临床开发。
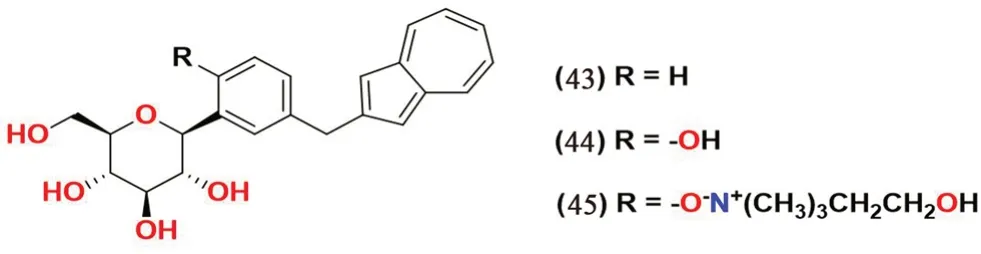
图11 薁类SGLT2 抑制剂化学结构Fig 11 Chemical structure of azulene SGLT2 inhibitors
2.3.9 异喹啉类(见图12) Pan 等[63]合成了一系列将四氢异喹啉环引入到C-芳基葡萄糖苷的化合物,并在体外测试了其对SGLT2 的抑制活性。经筛选发现化合物46 是最有效的SGLT2 抑制剂,可使SGLT2 活性降低81.7%,具有与达格列净(85.4%)相近的抑制作用。

图12 异喹啉类SGLT2 抑制剂化学结构Fig 12 Chemical structure of isoquinoline SGLT2 inhibitors
2.3.10 葡萄糖衍生物类(见图13) Chen 等[64]报道了在C-4 位具有双氟取代的达格列净衍生物的设计和相关合成,与参考化合物达格列净(IC50=1.98 nmol·L-1)相比,这一系列化合物在体外的SGLT2 抑制活性评估中表现出很强的抑制活性(IC50<0.60 nmol·L-1)。其中化合物47 是最有效的SGLT2 抑制剂,IC50值为0.35 nmol·L-1。SAR 分析表明吸电子基团(例如三氟甲基)的取代导致活性降低,而达格列净的C-4 位的双氟取代基则会明显提高SGLT2 抑制活性。
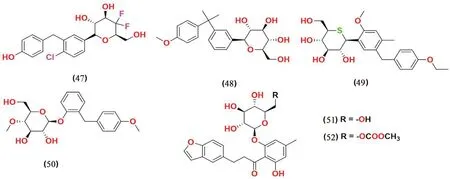
图13 葡萄糖衍生物类SGLT2 抑制剂化学结构Fig 13 Chemical structure of SGLT2 inhibitors of glucose derivatives
Wen 等[65]报道了二甲基取代的C-葡萄糖苷的合成,并对其进行SGLT2 抑制活性评估后得到了化合物48,在小鼠中显示出良好的降血糖功效,但其效力不如对照药物达格列净。
Kakinuma 等[66]报道了一系列将芳环或吡喃环连接到硫原子取代的D-葡萄糖的衍生物,并在体外测试了其对SGLT1 和SGLT2 的抑制活性,筛选出化合物49 为最有效的SGLT2 抑制剂,IC50值为2.26 nmol·L-1,选择性比SGLT1 强1650 倍。化合物49在接下来的体内测试中表现出较好的效果,目前已经进入临床Ⅱ期研究。
Cao 等[67]报道了一系列全新的葡萄糖苷化合物。通过优化,发现化合物50 具有良好的药代动力学稳定性和良好的药效学效力。
Tsujihara 等[68]设计并合成了一系列在B 环上取代的4'-脱羟基根皮苷的衍生物。研究发现,在4'位上有一个小的烷基取代基可以提高这些衍生物的生物活性。测试中化合物51 展现出了最优秀的抑制活性。对化合物51 的前药化合物52在小鼠中进行测试时观测到小鼠高血糖症的缓解和血糖水平的降低。
2.3.11 非糖类(见图14) Li 等[69]通过高通量筛选,得到了目标化合物53,使用中国仓鼠卵巢细胞实验进行体外测试时,发现其对SGLT2 的IC50值为1.4 μmol·L-1。通过对化合物53 修饰合成了一系列含有苯并噻嗪酮和苯并恶嗪酮结构的化合物,其中化合物54 和55 表现出较好的抑制活性。化合物54 对SGLT2 和SGLT1 的IC50值分别为0.009 μmol·L-1和9.14 μmol·L-1;化合物55 的IC50值分别为0.010 μmol·L-1和6.29 μmol·L-1。

图14 非糖类SGLT2 抑制剂化学结构Fig 14 Chemical structure of non-glucose SGLT2 inhibitors
3 结论
糖尿病及其并发症的危害较大,只有通过有效的方法控制血糖,才能减缓糖尿病的病情、减少并发症的出现。传统的降血糖药物只能在短期内控制患者的血糖,不能达到长期治疗的目的。SGLT2 抑制剂以其独特的作用机制区别于其他传统的降血糖药物,具有更好的治疗效果和更广阔的治疗范围。根皮苷是第一个被发现的具有SGLT 抑制活性的天然产物,但其选择性较差,因此,对其结构进行了大量的优化及改造,得到了许多选择性高、活性好、不良反应少的SGLT2 抑制剂。
随着对糖尿病深入的研究和探索,国内外越来越多的SGLT2 抑制剂进入临床研究中,如葛兰素史克公司的Remogliflozin[70]、Theracos 的Bexagliflozin[71]以及恒瑞公司的Henegliflozin[72]。与此同时,SGLT2 晶体结构的发现对新型选择性SGLT2 抑制剂的研究具有重要意义。
