γ-谷氨酰转肽酶高效表达及其催化合成γ-谷氨酰苯丙氨酸
2021-10-09刘栓英刘会灵龙梦飞武文慧张显徐美娟杨套伟饶志明
刘栓英,刘会灵,龙梦飞,武文慧,张显,徐美娟,杨套伟,饶志明
(江南大学 生物工程学院, 工业生物技术教育部重点实验室,江苏 无锡, 214122)
γ-谷氨酰转肽酶(γ-glutamyltranspeptidase,GGT)在临床诊断[1-2]、催化合成氨基酸衍生物[3]、药物或药物前体[4]以及改善苦味氨基酸的苦味等方面具有广泛应用[5]。研究表明氨基酸L-苯丙氨酸(L-phenylalanine,L-Phe)在食物中可以呈现出苦味[6],又是人体必需氨基酸[7]。SUZUKI等为改善其苦味,利用EscherichiacoliK-12来源的GGT 0.5 U/mL,以200 mmol/LL-谷氨酰胺(L-glutamine,L-Gln)为供体底物,200 mmol/LL-Phe为受体底物,最终γ-谷氨酰苯丙氨酸(γ-glutamylphenylalanine,γ-Glu-Phe)产率达到70%[6]。
Bacillussubtilis168(B.subtilis168)作为GGT表达的宿主细胞,存在分泌效率低、表达量不高的问题[8-9]。研究团队前期引入分子伴侣脂蛋白PrsA以及优化Poly(A/T)尾巴的长度,酶活力达到30.4 U/mL[10]。转录水平的高低对于基因表达量具有重要影响[11],启动子对于调节基因的转录水平发挥着重要的作用[12]。通过对-10区上游的保守区域进行非理性改造,可以提高目的蛋白表达量[13-14]。然而,目的基因在相同启动子的调控下,表达量也会受到起始翻译速率的影响[15]。核糖体结合位点(ribosome binding site,RBS)是影响起始翻译速率的重要生物学元件。通过优化编码目的基因的RBS序列,可使目标酶表达量提高10倍[16]。根据文献报道,RBS Calculator(https://www.denovodna.com/software/)可以计算得到具有不同起始翻译速率的RBS序列[17-18]。
传统方法获得高表达量的GGT菌株耗时且困难。本研究建立了一种基于显色反应的高通量筛选方法,能快速且准确筛选GGT高表达量菌株。通过对于启动子HpaII的-10区上游关键区域进行两轮迭代突变建立了启动子突变文库,经高通量筛选得到最优启动子。在此基础上,构建人工RBS序列文库,经筛选得到最优人工RBS序列,最大程度提高GGT表达量。另外,优化了GGT催化L-Phe生成γ-Glu-Phe的转化条件,提高了γ-Glu-Phe产率,从而为GGT应用于食品行业提供实验基础。
1 材料与方法
1.1 材料
1.1.1 菌株与质粒
枯草芽孢杆菌B.subtilis168、pMA5-ggt-prsA/B.subtilis168、大肠杆菌EscherichiacoliJM109、质粒pMA5由本实验室保藏。
1.1.2 酶和试剂
BamH I、MluI、DpnI限制性核酸内切酶、DNA聚合酶,TaKaRa公司;高保真酶、同源重组酶克隆试剂盒,南京诺维赞生物科技有限公司;琼脂糖凝胶DNA回收试剂盒、小量质粒提取试剂盒、细菌DNA基因组提取试剂盒,上海捷瑞生物工程有限公司;卡那霉素、氨苄霉素、γ-谷氨酰对硝基苯胺一水合物、双甘二肽、L-Phe、L-Gln,生工生物工程(上海)股份有限公司;γ-Glu-Phe标样、其他分析纯试剂,国药集团化学试剂有限公司。
1.1.3 培养基
LB液体培养基(g/L):酵母粉5.0,胰蛋白10.0,NaCl 10.0[19]。
LB固体培养基(g/L):酵母粉5.0,胰蛋白栋10.0,NaCl 10.0,琼脂粉20。
SpI以及SpII培养基:
SpA(g/L):K2HPO4·3H2O 28.0,KH2PO412.0,(NH4)2SO44.0,柠檬酸三钠2.0。
SpB(g/L):MgSO4·7H2O 0.4。
100×CAYE(g/L):酵母粉100.0,水解酪蛋白20.0, MgCl2·6H2O 5.08,CaCl25.5。
发酵培养基(g/L):葡萄糖10.0,酵母粉20.0,胰蛋白胨15.0,K2HPO4·3H2O 2.0,MgSO4·7H2O 1.0,pH 7.2。
1.2 实验方法
1.2.1 质粒提取和引物的设计
pMA5-ggt-prsA质粒的提取按照质粒提取试剂盒的方法进行。在本文中涉及到的引物如表1所示。

表1 本研究中使用的引物Table 1 Primers used in this study
1.2.2 构建启动子突变文库
以提取的质粒pMA5-ggt-prsA作为模板,以引物F1和R1进行PCR扩增,PCR产物在DpnI的作用下37 ℃消化1 h,将消化产物转化入B.subtilis168感受态细胞中,将重组细胞涂布在LB固体培养基(含50 μg/mL卡那霉素)上培养,菌落PCR筛选鉴定后,建立第一批启动子突变文库。在第一轮得到的正向启动子菌株中提取质粒,送至苏州金唯智生物科技有限公司测序,再以此质粒作为模板,以引物F2以及R2进行扩增,PCR产物在DpnI的作用下,37 ℃消化1 h,将消化产物转化入B.subtilis168感受态细胞中,将重组细胞涂布在LB固体培养基(含50 μg/mL卡那霉素)上培养,菌落PCR筛选鉴定后,建立起第二批启动子突变文库。
1.2.3 设计RBS序列文库
在得到最优启动子的基础上,以相应的质粒作为模板,以引物F3、F4和R3进行PCR,将人工RBS序列替换原始RBS,另外,以质粒pMA5为模板,利用引物F5和R4进行PCR,PCR产物在DpnI 的作用下,37 ℃消化1 h。将消化产物、改造后的ggt基因经核酸胶回收后,利用同源重组试剂盒进行连接,将连接产物转化入B.subtilis168感受态细胞中,涂布在LB固体培养基(含50 μg/mL卡那霉素)上培养,菌落PCR筛选鉴定后,建立起RBS文库。
1.2.4 高通量筛选操作流程
首先挑取1.2.2、1.2.3中的突变体文库中的阳性克隆子,在含有3 mL发酵培养基的96孔板中,37 ℃,200 r/min条件下培养48 h。离心获得含有GGT的发酵上清液,在含有50 mmol/LL-Phe、50 mmol/LL-Gln 的3 mL的发酵培养基中,在反应温度为37 ℃,用0.1 mol/L Tri-HCl控制反应pH为10.0,转速为200 r/min的条件下,反应10 min。取反应液10 μL与100 μL 20 mmol/L邻苯二甲醛(O-phthalaldehyde,OPA)工作液充分混合,振荡混匀20 min,然后先加入100 μL二甲基亚砜(dimethyl sulfoxide,DMSO),再加入50 μL 10%三氯乙酸(trichloroacetic acid,TCA)充分混匀10 min,利用酶标仪在600 nm处记录吸光值。
1.2.5 实时荧光定量PCR进行基因转录丰度分析
取培养好的细胞发酵液2 mL置于离心管中,12 000 r/min的条件下离心5 min,弃去上清液,菌体可利用诺唯赞试剂盒进行RNA提取。利用实时荧光定量PCR分析不同菌株GGT转录水平的具体步骤参考文献所述[10]。
1.2.6 酶的表达与纯化
取-40 ℃冰箱保存的菌种于固体LB平板划线活化,挑取单菌落接入含有卡那霉素(50 μg/mL)抗性的10 mL LB培养液中。37 ℃过夜培养后按照6%的接种量转接入含相同浓度抗生素50 mL发酵培养基中继续培养48 h,离心后获得含有目的蛋白的发酵液上清液。
首先配制pH 10.0的饱和硫酸铵溶液,以0%~20%,20%~40%,40%~60%,60%~80%,80%~100%饱和度硫酸铵梯度分级沉淀。取体积分数为40%的饱和硫酸铵沉淀的活性蛋白上清液,再以0%~20%,20%~40%,40%~60%,60%~80%,80%~100%饱和硫酸铵梯度沉淀,从而得到含有活性蛋白的沉淀,以Tri-HCl缓冲液(pH 8.0,0.1 mol/L)复溶,经透析后以Hitrap DEAE FF柱层析分离,上样缓冲液为Tri-HCl(pH 8.0,0.05 mol/L),洗脱液为Tri-HCl(pH 8.0,0.05 mol/L)+0.5 mol/L NaCl,以0%~100%B,30 min梯度洗脱,收集洗脱峰[20]。将得到的纯酶通过SDS-PAGE方法进行分析,以及采用Bradford法测定蛋白浓度。
1.2.7 酶活力的测定
取培养48 h的菌液50 mL,于4 ℃条件下10 000 r/min离心30 min收集含有活性目的蛋白的上清液。酶活力测定方法如下:1 mL的标准反应体系中包含100 mmol/L Tris-HCl(pH 10.0),2.5 mmol/L γ-L-谷氨酰-对硝基苯胺一水合物,60 mmol/L双甘二肽和5 μL适当稀释的酶液。37 ℃反应15 min,加入200 μL 3.5 mol/L的醋酸终止反应,以不添加双甘二肽,添加等体积的水的反应体系作为对照,利用酶标仪在410 nm处测定吸光度值。通过实验组和对照组的吸光度差值计算转肽酶活力。酶活力定义为:1 min 生成1 μmol的对硝基苯胺所需的酶量为1个酶活力单位。
1.2.8 γ-Glu-Phe的检测
以GGT作为催化剂,以L-Gln、L-Phe为底物,在适宜催化反应条件下进行转化,定时取样,每次取样过程中加入等体积的10%三氟乙酸终止转肽反应。得到的反应产物,通过0.22 μm过滤膜过滤。利用高效液相色谱仪1200 Infinity系列进行检测,检测条件为:紫外检测器,X BridgetM C18柱(Agilent 5 μm,4.6 mm×250 mm),流动相A为50 mmol/L乙酸钠(含1%的N,N-二甲基甲酰胺),流动相B为50%乙腈,流速1 mL/min,柱温40 ℃。
2 结果与分析
2.1 高通量筛选方法建立


表2 OPA与反应体系中化合物之间的显色反应Table 2 Chromogenic reactions between OPA and compounds present in the reaction system
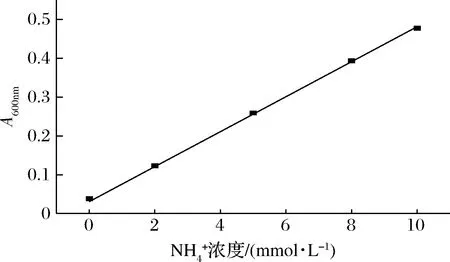
图1 释放的浓度和A600nm的关系Fig.1 The relationship between the concentration of released
2.2 通过优化启动子关键区域提高GGT表达量
HpaII是强组成型启动子,其-10区上游7个碱基是调控基因转录水平的关键区域。通过对关键区域进行非理性改造,提高基因的转录水平以及表达量。在得到的目的蛋白表达量提高的正向启动子突变中,通过实时荧光定量PCR、SDS-PAGE与酶活力检测等方式分析了基因的转录水平、蛋白表达以及酶活力情况。结果如图2、图3所示,较原始启动子HpaII相比,突变体EA13HpaII、EB6HpaII、ED11HpaII、EF3HpaII、EG2HpaII(基因序列见表3)启动子调控的基因转录水平明显提高,其中EF3HpaII的转录丰度较原始启动子HpaII提高了5倍。但是,检测pMA5-EF3HpaII-ggt-prsA/B.subtilis168酶活力较pMA5-HpaII-ggt-prsA/B.subtilis168(WT)仅提高了63%。这也就表明GGT的表达量不仅仅和基因的转录水平相关。
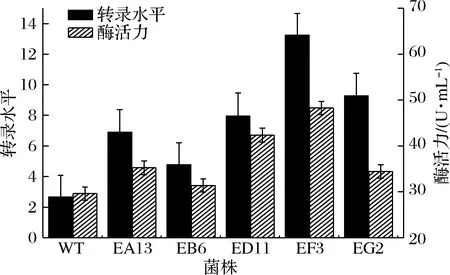
图2 不同启动子菌株中GGT的表达量以及转录丰度Fig.2 Transcription level and GGT production of recombinant strains with different promoters注:GGT 以HpaII(WT)、EA13HpaII、 EB6HpaII、ED11HpaII、EF3HpaII及EG2HpaII为启动子进行表达
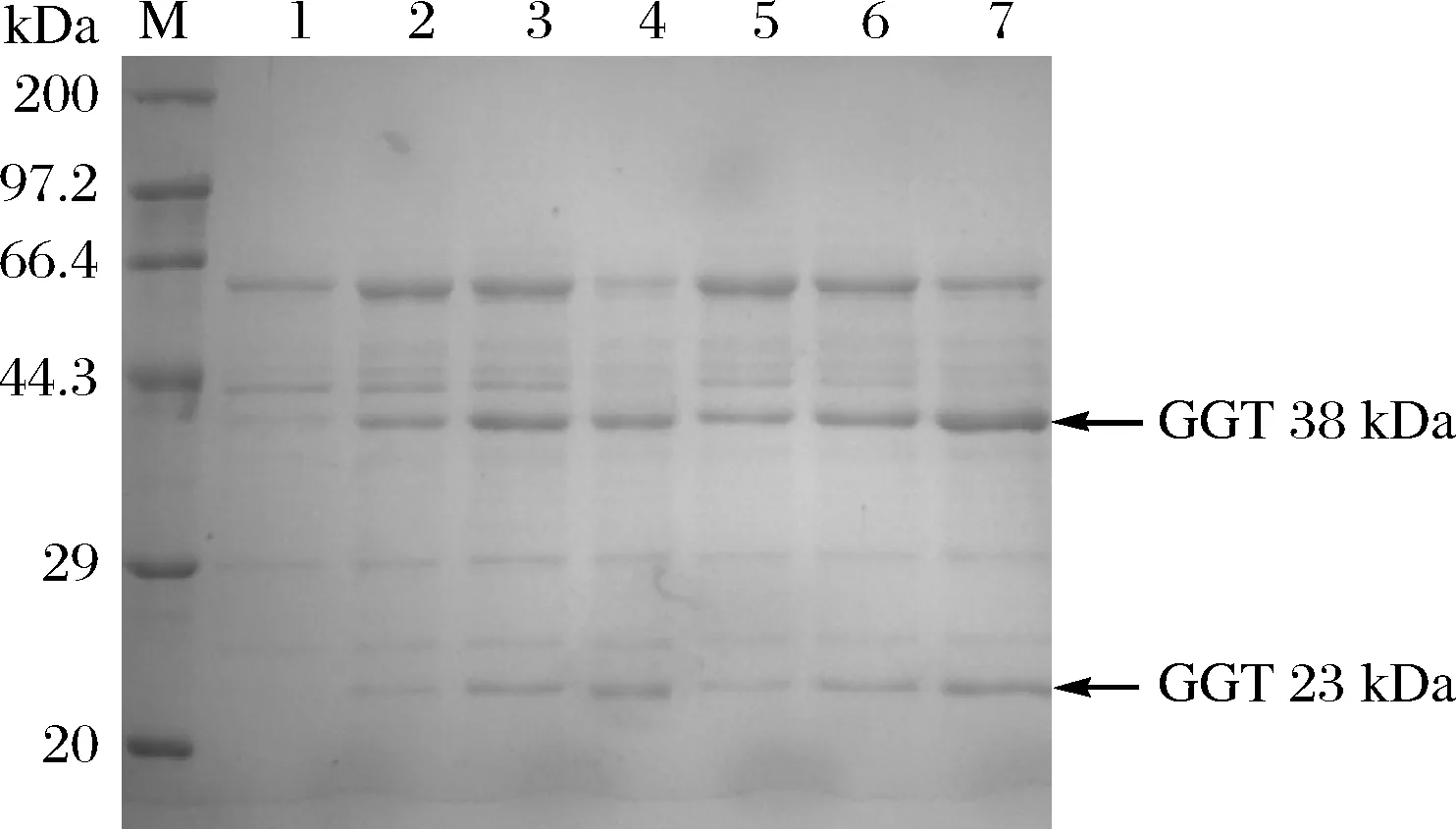
M-Marker;1-阴性对照,B.subtilis 168/pMA5;2~7-GGT在B.subtilis 168中以 HpaII、ED11HpaII、EB6HpaII、EG2HpaII、EA13HpaII及EF3HpaII为启动子进行表达图3 SDS-PAGE分析不同启动子重组菌株中GGT的表达量Fig.3 SDS-PAGE analysis of GGT production in recombinant strains with different promoters.
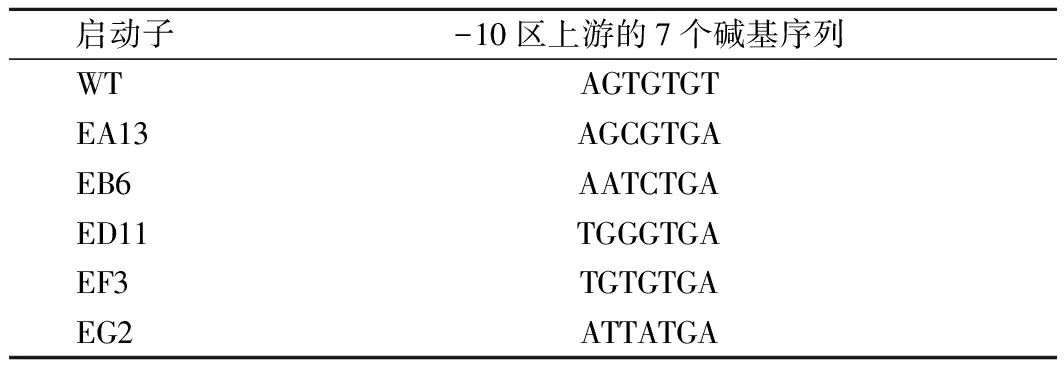
表3 不同启动子-10区上游的7个碱基序列Table 3 The 7 bases upstream of the -10 region in different promoters
2.3 RBS序列对于ggt表达的影响
为了探究2.2中ggt基因转录水平与蛋白表达提高程度存在重大差异的原因,首先,我们用DNAMAN软件分析了基因起始密码子前40 bp左右的EF3HpaIIRBS序列,在EF3HpaIIRBS的SD序列中,存在着多个明显的颈环二级结构,这严重影响基因的起始翻译速率,更有甚者导致基因无法表达[21-22]。
另外,通过RBS Calculator分析EA13HpaII、EB6HpaII、ED11HpaII、EF3HpaII、EG2HpaII以及HpaII起始翻译速率(表4),HpaII的起始翻译速率明显低于EF3HpaII等其他正向启动子。通过以上结果,表明基因的起始翻译速率可能就是造成基因转录水平与蛋白表达提高程度不一致的原因。因此,找到与正向启动子更加适配的RBS序列是进一步提高目的基因表达量的关键。
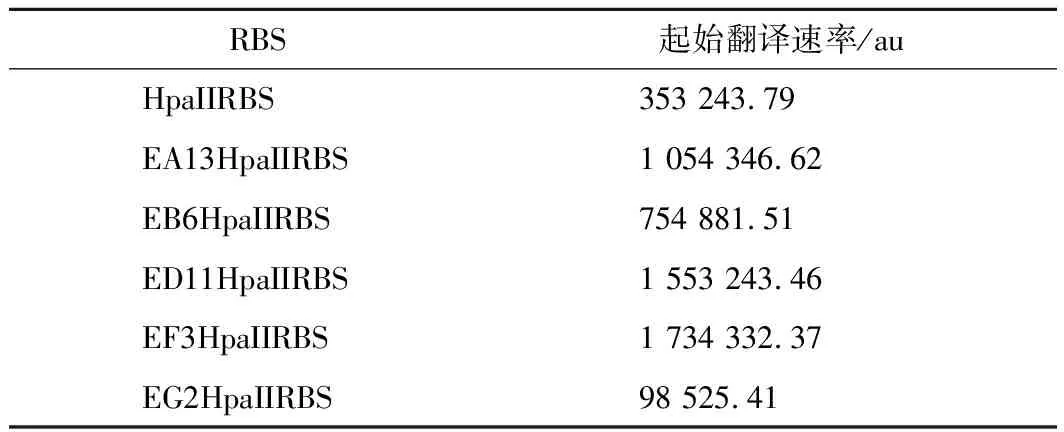
表4 不同RBS序列的起始翻译速率Table 4 The initial translation speeds of different RBS sequences
2.4 优化ggt起始翻译速率提高表达量
为了进一步提高GGT的表达量,我们利用RBS Calculator设计与启动子正向突变菌株pMA5-EF3HpaII-ggt-prsA/B.subtilis168更加适配的RBS序列,从而得到具有梯度起始翻译速率(1 000~4 986 270.7 au)的500个RBS序列文库。按照其起始翻译速率不同,将其共分为10组,1×103~2×103、2×103~5×103、5×103~1×104、1×104~5×104、5×104~1×105、1×105~2×105、2×105~5×105、5×105~1×106、1×106~2×106、2×106~4 986 270.7 au,每组50个RBS。
最终,我们得到362个较EF3HpaII原始RBS酶活力提高的人工RBS序列。根据不同RBS序列调控表达的GGT的转肽酶活力差异,从低到高排列为RBS1~RBS362,其中RBS361、RBS362起始翻译速率为2 986 410.3和3 974 351.5 au。结果如图4所示,重组菌株RBS361和RBS362经培养表达后,转肽酶活力为56.95和60.25 U/mL,是菌株pMA5-HpaII-ggt-prsA/B.subtilis168(WT)的1.91和2.02倍。基于此,我们又利用生物分析软件DNAMAN分析了RBS361和RBS362的SD序列,结果如图5所示,经优化后的人工RBS361和RBS362的颈环二级结构得到明显改善。以上研究结果表明,在菌株pMA5-EF3HpaII-ggt-prsA/B.subtilis168中,人工RBS序列比原始RBS序列更适配,从而能更大程度上提高GGT的表达量。
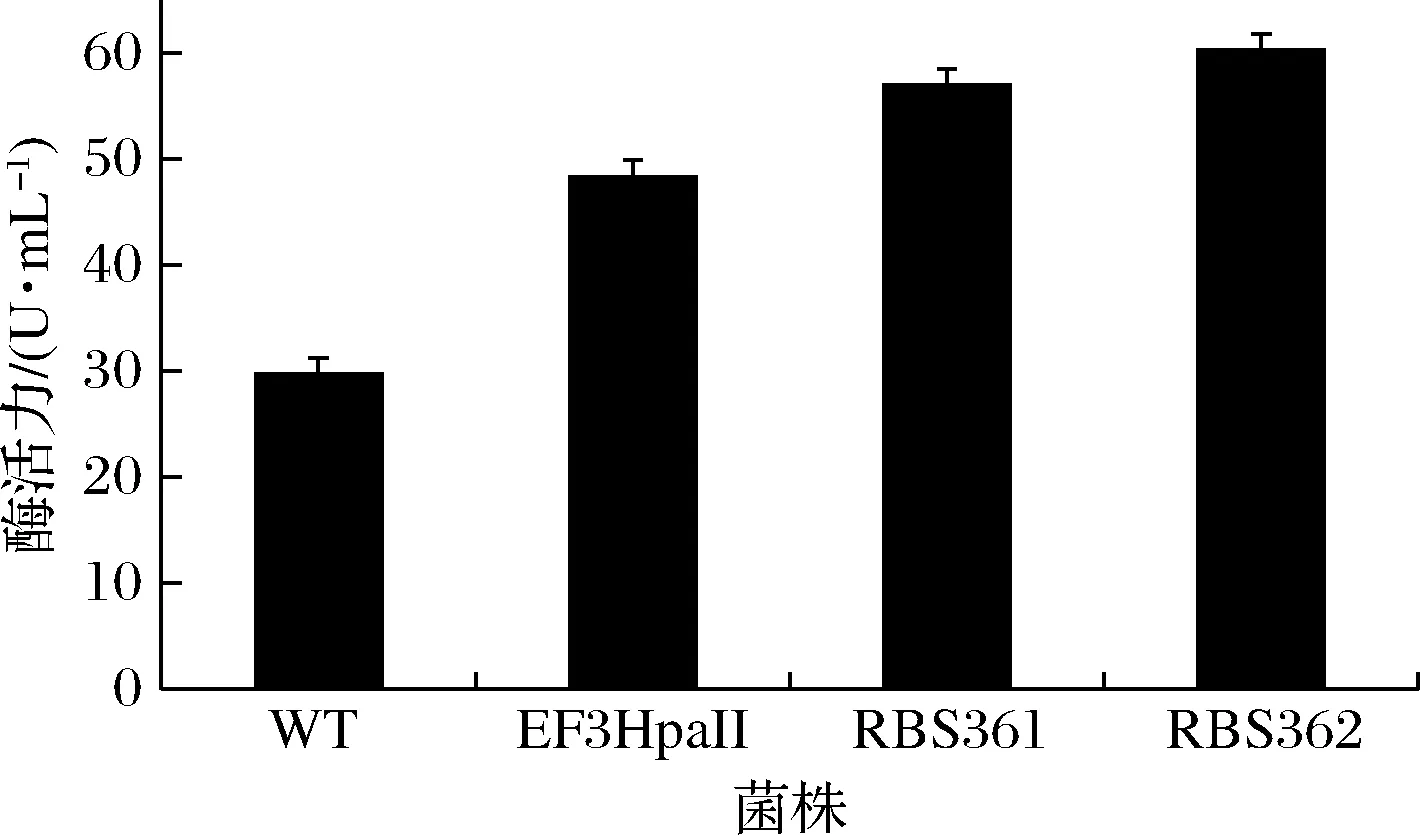
图4 不同RBS序列重组菌株GGT的表达量Fig.4 GGT production of recombinant strains with different RBS sequencesGGT在HpaII(WT)、EF3HpaII、EF3HpaIIRBS361及EF3HpaIIRBS362调控下进行表达
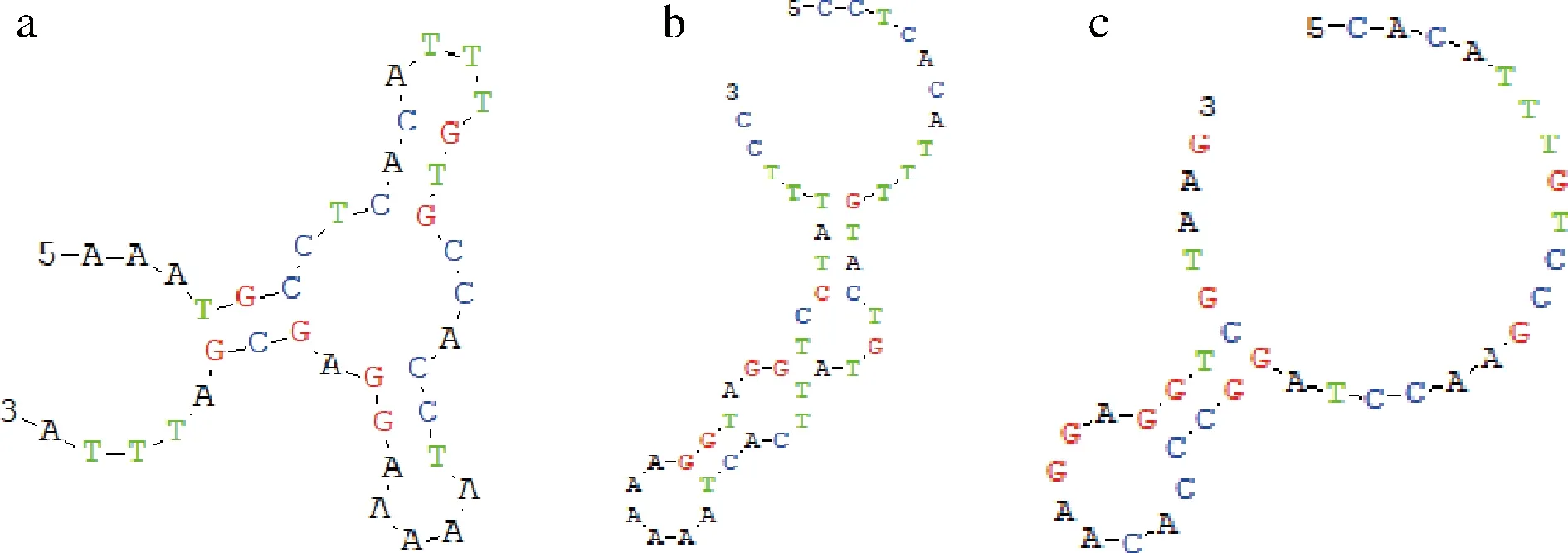
a-EF3HpaII RBS;b-EF3HpaII RBS361;c-EF3HpaII RBS362图5 不同RBS中SD序列的二级结构Fig.5 The secondary structure of SD sequences in different RBS
2.5 GGT催化L-Phe合成γ-Glu-Phe的条件优化
在前期的研究中,我们已经对于GGT的酶学性质进行了探索[10]。本研究在GGT催化合成γ-Glu-Phe的催化反应过程中,优化了反应体系中底物浓度配比、反应温度、初始反应pH以及酶量等催化条件,提高了γ-Glu-Phe产率。
2.5.1 底物浓度对GGT催化合成γ-Glu-Phe的影响
由于提高受体浓度有利于降低GGT水解副反应,优化底物浓度配比可以提高γ-Glu-Phe产率。控制反应温度为40 ℃,反应体系pH 10.0,其他催化条件不变,控制供体底物50 mmol/LL-Gln与受体底物L-Phe浓度配比为1∶1、1∶2、1∶3、1∶4、1∶5、1∶6,进行体外转化。其结果如图6-a所示,当供体底物50 mmol/LL-Gln与受体底物L-Phe浓度配比为1∶4时,产物产量达到最高,产量为10.62 g/L,产率达到70%。
2.5.2 温度对GGT催化合成γ-Glu-Phe的影响
温度可以影响酶促反应速率,从而影响酶催化合成γ-Glu-Phe。控制供体底物50 mmol/LL-Gln与受体底物L-Phe浓度配比为1∶4,反应体系pH 10.0,其他催化条件一致的情况下,分别在25、30、35、40、45 ℃ 下进行酶催化反应合成γ-Glu-Phe产物。结果如图6-b所示,当反应温度为35 ℃时,产物产量最高,达到11.98 g/L,产率达到79%。
2.5.3 初始pH对GGT催化合成γ-Glu-Phe的影响
控制供体底物50 mmol/LL-Gln与受体底物L-Phe 浓度配比为1∶4,反应温度为35 ℃,其他催化条件一致的情况下,设置转化体系初始pH 6.0、7.0、8.0、9.0、10.0、11.0、12.0条件下进行酶催化反应合成γ-Glu-Phe产物。结果如图6-c所示,在初始反应pH<10.0时,产物产率随pH的升高而升高。当pH为10.0左右时,产物产量达到最高,产量为12.14 g/L,产率为80%,在初始反应pH>10.0时,产物产量大幅度下降。这是由于GGT最适转肽酶活力pH=10.0,当pH>10.0时,GGT转肽活性降低[10],从而其催化合成产物γ-glu-Phe的能力下降,导致产物产率下降。
2.5.4 酶浓度对于GGT催化合成γ-Glu-Phe的影响
实际工业化应用中,为了最大程度提高生产效率和降低生产成本,将催化反应体系中酶浓度控制在适宜的水平也是至关重要的。控制其他催化条件相同,采用供体底物50 mmol/LL-Gln与受体底物200 mmol/LL-Phe,将反应温度设为35 ℃,反应初始pH 10.0,酶浓度分别为0.2、0.6、1、1.4、1.8 U/mL 条件下进行酶催化反应合成γ-Glu-Phe产物。结果如图6-d所示,当反应体系中酶浓度<1 U/mL时,底物转化率以及产物产率随酶浓度增加而逐渐升高,当酶量在1 U/mL左右时,产物产量达到最高,产量为14.35 g/L,产率达到94%。当GGT浓度过高时,会发生自转肽反应,从而生成谷氨酸等副产物[3]。所以,当酶量>1 U/mL时,γ-Glu-Phe产物产率大幅度下降。
3 结论与讨论

