胰岛素信号通路在运动改善阿尔茨海默病中的作用机制
2021-09-29任鹏飞
任鹏飞
阿尔茨海默病(Alzheimer's Disease,AD)是一种神经系统退行性疾病,是老年痴呆中最常见的一种疾病。2017年,全球约有4 400万AD患者,到2050年,全球AD患者可能达到1.15亿[1]。AD主要临床表现为认知功能障碍和记忆力衰退,其病理学特征为β-淀粉样蛋白(β-amyloid,Aβ)沉积、Tau蛋白过度磷酸化、神经元丢失和神经元内颗粒空泡变性,此外还包括糖代谢异常以及氧化应激增加等[2]。人体研究发现,外周胰岛素抵抗会增加AD发病机率,且大部分AD患者存在胰岛素抵抗和胰岛素分泌缺陷,因而AD也被认为是一种“脑型糖尿病”[3]。此外,动物实验也表明,AD转基因小鼠脑内存在胰岛素抵抗[4]。而抑制AD脑内胰岛素抵抗可以显著改善AD病理表现[5]。胰岛素信号通路受损是出现脑部胰岛素抵抗的重要原因,因而改善AD脑内胰岛素信号通路受损,可能是缓解AD的有效途径。其中,磷脂酰肌醇-3-激酶/丝苏氨酸激酶(Phosphatidylinositol-3-Kinases/Serine Threonine Kinase,PI3K/AKT)、丝裂原活化蛋白酶(Mitogen-Activated Protein Kinase,MAPK)、Wnt通路是3条主要的胰岛素信号通路[6]。
已有研究证明,运动可以减少AD脑内Aβ沉积,抑制Tau蛋白过度磷酸化,改善细胞自噬水平,显著提高学习记忆能力,但运动改善AD的作用机制仍不明晰。现有文献表明,运动可能通过激活PI3K/AKT、MAPK和Wnt信号通路,改善AD病理。因此,本文将从上述3条胰岛素信号通路切入,进行研究综述。
1 PI3K/AKT信号通路与AD
PI3K/AKT信号通路受胰岛素、神经生长因子等调控,参与细胞增殖、分化、存活等诸多生物学功能。激活后的PI3K和AKT的PH结构域结合,改变构象并发生位移,磷酸化AKT的Ser473和Thr308位点从而激活AKT,活化的AKT作用于糖原合成酶3(Glycogen Synthase Kinase3,GSK3)和哺乳动物雷帕霉素靶蛋白(Mammalian Target of Rapamycin,mTOR)等下游因子,调节细胞的生长和凋亡[7]。目前研究已经证实,PI3K/AKT信号通路可能通过调控Tau蛋白、Aβ、神经炎症和自噬,调控AD病理特征。
1.1 运动激活PI3K/AKT减少Aβ沉积
Aβ是由β-淀粉样前体蛋白(β-Amyloid Protein Precursor,APP)被β-分解酶和γ-分解酶水解后生成的。正常情况下,脑内Aβ的生成与降解存在动态平衡,在AD中这种平衡被打破,脑内Aβ大量聚集。研究显示,PI3K/AKT信号活性降低时,Aβ出现过度沉积[8]。中药清心开窍方可激活APP/PS1转基因小鼠海马组织PI3K/AKT信号通路,降低Aβ水平[9]。且激活PI3K/AKT信号通路可通过降低β-分泌酶活性减少Aβ含量[10]。进一步研究证实,降低PI3K/AKT信号通路活性可以增加糖原合成激酶-3α(Glycogen Synthase Kinase-3α,GSK-3α)磷酸化,提高β-分泌酶活性,促进Aβ40和Aβ42生成。同时,Aβ40和Aβ42可通过增加Ca2+进入神经元,激活GSK-3β和细胞周期素依赖蛋白激酶5(Cyclin-Dependent Kinase5,CDK5),进一步阻断PI3K/AKT通路活性,最终导致Aβ过度沉积和AD认知功能障碍[11]。提示增加PI3K/AKT信号通路活性可以通过降低GSK-3α磷酸化水平,降低β-分泌酶,抑制AD脑内Aβ生成。
有研究报道,5个月跑台运动降低了APP/PS1小鼠海马组织中GSK-3α的Ser21位点磷酸化水平,抑制了Aβ沉积[12]。6周跑台运动可以提高糖尿病鼠海马内AKT表达,降低GSK-3α磷酸化,减少Aβ沉积,改善认知功能[13]。提示运动可能通过改善PI3K/AKT信号通路活性,降低GSK-3α磷酸化水平,减少Aβ沉积。另有研究发现,8周有氧运动激活了小鼠皮质和海马组织中PI3K/AKT信号通路,降低了β-分泌酶水平,减少了Aβ42表达,但APP蛋白含量并没有发生显著改变[14]。提示有氧运动通过激活PI3K/AKT信号通路,抑制β-分泌酶产生,使APP蛋白向非Aβ途径转移,产生可溶的小分子蛋白,减少Aβ生成。以上研究说明,运动可以激活PI3K/AKT信号通路,降低GSK-3α磷酸化水平,减少β-分泌酶含量,从而抑制Aβ生成,改善AD认知能力。
1.2 运动激活PI3K/AKT改善Tau蛋白过度磷酸化
Tau蛋白异常磷酸化是AD主要病理特征之一。对AD患者尸检发现,颞叶皮层中过度磷酸化的Tau蛋白显著增多,同时伴随有磷酸化AKT水平显著降低[15]。Schubert等[16]也指出,抑制PI3K/AKT信号通路会导致Tau蛋白过度磷酸化。而激活PI3K/AKT信号通路可减少Tau蛋白的Ser404和THr231位点磷酸化水平,进而改善AD。提示,激活PI3K/AKT信号通路可以改善Tau蛋白过度磷酸化。Tanabe等[17]的研究发现,在Tau蛋白过度磷酸化的AD模型鼠脑中,AKT活性下降且伴随着GSK-3β活性增加。Eph-B2受体可通过激活PI3K/AKT信号通路,抑制GSK-3β活性,改善Tau蛋白过度磷酸化[18]。上述研究提示,PI3K/AKT信号通路可以通过抑制GSK-3β活性,改善Tau蛋白过度磷酸化。
研究发现急性有氧运动后短期内大鼠海马组织中PI3K/AKT信号通路活性增加,GSK-3βSer9位点磷酸化水平升高,GSK-3β活性下降,其下游底物Tau蛋白的磷酸化水平也显著降低;而48 h后,大鼠海马组织中AKT和GSK-3β的磷酸化水平降低,Tau蛋白磷酸化恢复至基线水平[19]。提示急性运动后短期内激活PI3K/AKT信号通路可改善Tau蛋白的过度磷酸化,但具有时效性。长期运动训练也可以激活PI3K/AKT信号通路,改善AD病理。3个月耐力运动上调了AD小鼠脑内PI3K和AKT的磷酸化表达,降低了AD小鼠海马组织Tau蛋白磷酸化水平[20]。 房国梁等[21]发现,8周抗阻运动提高了大鼠大脑皮质和海马组织PI3K的含量及活性,促进了AKT在Thr308和Ser473位点的磷酸化,活化的AKT通过磷酸化GSK-3βSer9位点,抑制GSK-3β活性,抑制了Tau蛋白多个位点的过度磷酸化。以上研究均说明,运动可以通过激活PI3K/AKT信号通路,促进GSK-3β磷酸化,抑制GSK-3β活性,缓解Tau蛋白过度磷酸化,改善AD认知功能障碍。
1.3 运动激活PI3K/AKT改善神经炎症
已有研究证实,AD转基因小鼠脑内存在胰岛素抵抗,胰岛素抵抗可导致机体巨噬细胞、平滑肌细胞等产生大量炎症因子,如白介素-1(Interleukin-1,IL-1)、核转录因子κB(Nuclear FactorκB,NF-κB)和肿瘤坏死因子-α(Tumor Necrosis Factor-α,TNF-α)等,发生炎症反应。毛小元等[22]研究发现,应用蛇床子素(OST)治疗糖尿病脑病大鼠后,降低了大鼠海马组织中炎症因子NF-κB、TNF-α和IL-1β,加入PI3K抑制剂后,加重了炎症反应,提示炎症反应与PI3K/AKT信号通路有关。硝唑尼特(Nitazoxanide,NTZ)处理LPS诱导的BV2细胞后,激活了PI3K和AKT,增加了IκB表达,并抑制了NF-κB的核易位[23]。表明NTZ降低炎症反应是通过PI3K/AKT/IκB/NF-κB途径实现的。同时,NTZ治疗APP/PS1小鼠后,小鼠脑中促炎因子IL-1β、TNF-α和诱导型一氧化氮合酶(Inducible Nitric Oxide Synthase,iNOS)表达显著降低,提示NTZ可能通过PI3K/AKT信号通路降低小鼠脑中的促炎因子。
研究表明,游泳运动激活了D-半乳糖诱导AD大鼠海马组织中IGFI-R/PI3K/AKT信号通路,且降低了炎症因子TNF-α、p-NF-κB、环氧化酶(Cyclooxygenase,COX-2)和iNOS的蛋白水平,提示运动可能通过激活PI3K/AKT信号通路,降低海马组织内的炎症反应[24]。另有Wang等[25]研究发现,有氧运动激活了糖尿病大鼠前额叶中PI3K/AKT信号通路,增加了下游因子FOXO1的磷酸化,降低FOXO1乙酰化水平及活性,抑制其下游NF-κB表达,从而抑制炎症反应。
1.4 运动激活PI3K/AKT增加自噬水平
PI3K/AKT信号通路下游的靶蛋白mTOR是调节自噬的主要因子,其主要复合物mTOR复合物1(mTOR complex1,mTORC1)参与调节细胞生长、凋亡、能量代谢和细胞自噬,活化的mTORC1可以抑制自噬并促进神经元中的蛋白质合成。不同刺激物(例如胰岛素、IGF、生长因子和氨基酸等)可以激活PI3K-AKT信号通路,通过结节性硬化症蛋白复合体(Tuberous Sclerosis Complex,TSC)抑制mTORC1上游信号蛋白Rheb,从而抑制mTORC1生成,进而诱导自噬过程。有研究表明mTOR诱导的自噬活动与AD 2种病理过程(Aβ沉积和Tau蛋白过度磷酸化)密切相关。研究发现,Aβ沉积加剧mTOR信号传导,而抑制mTOR信号可降低Aβ水平[26]。此外,Shen等[27]发现,应用mTOR抑制剂抑制mTOR活性可改善Tau蛋白过度磷酸化。因此,激活PI3K/AKT信号通路可以抑制下游分子mTOR表达,调控AD脑内自噬,减少Aβ沉积和Tau蛋白过度磷酸化,改善AD认知。
先前研究指出,12周跑台运动激活了自噬水平,减少了APP/PS1转基因小鼠海马组织中Aβ沉积[28]。同时,运动可以通过激活PI3K/AKT信号通路,缓解AD病理[29]。推测运动可通过激活PI3K/AKT通路激活自噬,促进Aβ清除。Kang等[30]研究发现,NSE/htau23转基因AD小鼠大脑皮层中mTOR磷酸化异常,自噬标志性蛋白Beclin-1和LC3B减少,自噬受损,而12周跑台运动激活了PI3K/AKT信号通路,抑制其下游分子mTOR活性,增加自噬蛋白Beclin-1表达,增强自噬活性,延缓AD小鼠认知功能下降。因此,运动可激活PI3K/AKT通路,抑制mTOR活性,增加AD脑内自噬水平,缓解AD症状。
综上,运动可以激活PI3K/AKT信号通路调控下游分子GSK-3β、GSK-3α、NF-κB和mTOR等,从而降低Tau蛋白过度磷酸化、减少Aβ沉积、缓解炎症反应和增强自噬水平,改善AD认知能力。
2 MAPK信号通路与AD
MAPK信号通路是哺乳动物细胞中的重要胰岛素信号通路。MAPK信号通路可以被生长因子、细胞因子、激素和细胞应激等细胞外刺激激活,继而触发三级酶促级联反应(MAPKKK-MAPKK-MAPK),参与细胞生长、分化和凋亡等生理过程[31]。目前在哺乳动物细胞中发现3条MAPK信号通路:细胞外信号调节蛋白激酶(Extracellular Signal-regulated Kinase,ERK)、c-Jun N末端激酶(c-Jun N-terminal Kinase,JNK)和p38丝裂原活化蛋白激酶(p38 Mitogen Activated Protein Kinases,p38MAPK),这3条通路与AD特征性病理改变有密切关系。
2.1 运动调控MAPK减少Aβ沉积
p38MAPK是MAPK家族中介导细胞存活和凋亡的重要通路。研究发现,AD小鼠脑内p38MAPK活化水平显著升高[32]。敲除AD模型小鼠p38αMAPK的编码基因mapk14,降低p38αMAPK表达,发现β-分泌酶1(β-site APP cleaving enzyme1,BACE1)溶酶体降解,BACE1表达降低,AD小鼠脑内Aβ生成减少。提示抑制p38MAPK表达,可降低BACE1表达和活性,抑制Aβ生成。除了p38MAPK信号通路,细胞外调节蛋白激酶 (Extracellular Regulated Protein Kinase,ERK)信号通路也是MAPK经典信号转导通路。AD鼠海马组织内,ERK磷酸化水平降低,Aβ沉积增加[33]。而增加APP/PS1小鼠海马组织中ERK磷酸化水平,可以抑制APP和BACE1基因表达,减少Aβ沉积[34]。提示提高ERK信号通路活性可通过抑制APP和BACE1基因表达,减少Aβ生成。JNK是MAPK的一种亚类,又被称为应激活化蛋白激酶(SAPK),近期有研究发现,c-Jun氨基末端激酶(c-Jun Nterminal Kinase,JNK)信号通路与AD发病密切相关,参与早期老年斑形成[35]。Savage等[36]用特异性活化抗体检测不同基因鼠中的MAPK信号通路,证实激活JNK信号通路可导致Aβ沉积增加。喂养雄性TgCRND8转基因小鼠4个月异丁香胆碱(IRN)抑制JNK信号通路后发现,Aβ沉积减少,小鼠认知功能得到改善[37]。 综上,在AD中,抑制p38MAPK和JNK信号通路,激活ERK信号通路可减少Aβ沉积。
研究发现,跑台运动抑制了AD模型鼠脑内MAPK通路(p38和JNK),提高了认知功能[38]。此外,急性跑台运动上调了高脂膳食诱导肥胖小鼠脑内ERK的表达,降低了BACE1活性,减少了Aβ沉积,而JNK和p38MAPK表达未有显著改变[39]。提示,急性跑台运动可以通过激活ERK通路,减少Aβ沉积,但急性运动对JNK和p38MAPK通路无显著作用,可能受长期运动影响才能实现对Aβ清除。而Pena等[40]发现,9周抗阻训练降低了3xTg-AD小鼠海马组织中APP水平,但未改变ERK蛋白含量及磷酸化水平。提示,运动激活ERK信号通路减少Aβ沉积,可能与运动类型有关。综上,未来仍需进一步探究不同运动类型及运动周期干预MAPK(JNK、p38MAPK、ERK)信号通路从而减少Aβ沉积的作用机制。
2.2 运动调控MAPK改善Tau蛋白过度磷酸化
Tau蛋白Thr231位点过度磷酸化可激活MAPK信号通路,促进神经元中细胞周期激活机制,导致细胞死亡[41]。同时,JNK、p38和ERK等几种激酶也可使Tau蛋白过度磷酸化。APP二聚化可激活ASK1-MKK6-p38级联反应,使Tau过度磷酸化,导致认知功能障碍[42]。GSK-3是Tau蛋白磷酸化的重要激酶,有研究发现,下调B103-695细胞中APP表达可激活Ras-ERK信号通路,抑制GSK-3活性,降低Tau蛋白过度磷酸化[43]。提示激活ERK信号通路,可降低Tau蛋白过度磷酸化。另有研究发现,药物利拉鲁肽治疗AD转基因小鼠后,激活了ERK信号,抑制了JNK表达,改善了Tau蛋白高磷酸化水平[44]。以上研究说明,抑制p38MAPK、JNK通路和激活ERK通路,可改善Tau过度磷酸化,进而调控AD认知障碍。
运动可以改善AD脑内Tau蛋白过度磷酸化,其机制可能与海马组织中的MAPK信号通路有关。Wang等[45]发现,12周跑台运动降低了JNK和p38MAPK磷酸化水平,提高了ERK1/2磷酸化水平,减少了Tau蛋白过度磷酸化,改善了AD转基因小鼠认知功能障碍。另外,12周跑台运动增加了24月龄Tg-NSE/PS2m AD小鼠海马组织内ERK磷酸化水平,降低了JNK和p38MAPK磷酸化水平,降低了Tau蛋白在Ser404、Ser202和Thr231位点的磷酸化水平[46]。提示运动通过抑制JNK、p38MAPK信号通路和激活ERK信号通路,降低Tau蛋白磷酸化,从而改善AD认知功能。
2.3 运动调控MAPK改善神经炎症
AD中存在神经炎症,且与MAPK信号通路有关。研究发现,Vitegnoside抑制了p38MAPK和JNK信号通路,减少了下游NF-κB炎症信号转导,缓解了AD细胞模型中的炎症反应[47]。另有研究发现,中药苓桂术甘汤治疗AD大鼠显著抑制了AD大脑中p38表达,上调了ERK信号,降低了NF-κB和IκBα表达,减少了促炎细胞因子产生[48]。以上研究说明,抑制p38MAPK和JNK信号通路,激活ERK信号通路,可降低下游炎症信号NF-κB转导,减少炎症因子表达,缓解AD神经炎症。
研究发现,跑台运动下调了早老素2突变小鼠海马组织中的ATF6α和sXBP1的表达,抑制了小鼠海马组织中JNK和p38MAPK信号通路,降低了海马组织中炎症因子TNFα和IL-1α表达,改善了小鼠认知障碍[49]。另有实验发现,运动抑制了AD模型鼠海马组织中JNK和p38MAPK信号通路,激活了ERK信号通路,降低了海马中TNF-α和IL-1β水平[38]。因此,运动可通过抑制海马组织中的JNK和p38MAPK信号通路,激活ERK信号通路来降低炎症反应。
2.4 运动调控MAPK改善自噬水平
脂多糖(Lipopolysaccharide,LPS)降低了小胶质细胞BV2中自噬标记物LC3-II的表达,并且LPS诱导了p38αMAPK激活,而SB203580(p38αMAPK抑制剂)处理后,发现LPS诱导的p38αMAPK活化被抑制,LC3-II表达显著上升[50]。提示抑制p38MAPK信号通路会提高自噬水平。在AD小鼠脑内皮层和海马神经元中特异缺失p38αMAPK,LC3-II和Beclin-1表达显著升高,提高了AD小鼠脑内自噬水平[51]。综上,可通过抑制p38MAPK通路,增加下游相关自噬因子LC3-II和Beclin-1表达,增加AD内自噬水平。
研究发现,递增负荷训练下调了衰老小鼠中转化 生 长 因 子-β1(Transforming Growth Factor-β1,TGF-β1)及其下游信号分子TGF-β活化激酶1(TAK1)、MAPK、MKK3和p38MAPK的 表 达,且E-cadherin,Beclin-1和LC3的蛋白表达水平显著增加[52],提示递增负荷训练可通过抑制TGF-β1/TAK1/MMK3/p38MAPK信号通路,激活自噬水平,从而改善老年小鼠的肾纤维化。因此,猜测运动可能会通过抑制p38MAPK信号通路提高AD自噬水平,改善AD认知障碍。目前尚无运动干预AD模型调控MAPK信号通路改善自噬的相关研究,未来该方向有待进一步研究。
3 Wnt信号通路与AD
Wnt信号通路在神经元分化、迁移、突触发生等过程中起关键作用。其中,Wnt/β-连环蛋白信号通路(canonical Wnt/β-catenin)作 用 最 为 广 泛,该 通 路中,Wnt蛋白是重要的启动因子,当Wnt蛋白不存在时,Wnt/β-catenin信号通路被抑制,胞内的支架蛋白Axin、APC与GSK-3β和Ⅰ型酪蛋白激酶(Caseinkinase1,CK1)形成破坏复合物,介导β-catenin磷酸化,泛素介导的蛋白酶体识别并降解β-catenin,导致Wnt/β-catenin途径中断。 当Wnt蛋白存在时,Wnt/β-catenin信号通路被激活,Wnt与细胞表面受体(Frizzled)和膜辅助受体-低密度脂蛋白受体相关蛋白(LRP5/6)结合,使散乱蛋白与Axin结合,破坏Axin/APC/GSK-3β/CK1形成的复合物,抑制β-catenin的磷酸化[53]。β-catenin进入细胞核并与转录因子(Lymphoid Enhancer-Binding Factor 1,LEF1)/T细胞因子(T Cell Factor,TCF)结合,引起Wnt靶基因的转录,参与生物体的生长、发育以及分化等活动。研究发现,Wnt/β-catenin信号通路与多种神经系统疾病的发生和发展有关,尤其是与AD的发病密切相关。Wnt/β-catenin信号通路可调控Aβ的生成与清除、Tau蛋白过度磷酸化、神经炎症以及自噬等影响认知功能。
3.1 Wnt与Aβ
在AD患者及动物模型中,Aβ的生成和清除与Wnt/β-catenin信号通路有关。CHO 7PA2细胞实验发现,抑制Wnt/β-catenin信号通路,Aβ42浓度增加,Aβ42/Aβ40比率和低分子量Aβ低聚物(如三聚体和四聚体)增加,提示阻断Wnt信号可导致APP发生淀粉样蛋白水解,增加C99和Aβ42表达[54]。动物实验发现,抑制J20TgAD小鼠Wnt/β-catenin信号通路,增加了小鼠海马组织Aβ42产生,小鼠记忆力受损[55]。进一步研究发现,在Wnt/β-catenin信号通路中,Aβ可与GSK-3β结合,降解β-catenin引起神经细胞毒性,加重认知障碍[56]。同时,脑内沉积的Aβ又可通过上调Dickkopf1(DKK1)表达抑制Wnt/β-catenin信号通路,导致Aβ清除速率进一步降低,加重AD病情[57]。在APP/Tau/PS1小鼠中,通过氟西汀增加β-catenin含量、抑制GSK-3β活性,可以减少APP裂解及Aβ生成,起到神经保护作用[58]。由此可知,激活Wnt信号通路可抑制Aβ产生,改善AD认知功能障碍。
3.2 Wnt与Tau蛋白
在J20TgAD小鼠中,Wnt信号功能障碍会加速Tau蛋白位点Thr231,Ser235和AT8的磷酸化,且发现阻断Wnt途径的野生型小鼠中也存在Tau蛋白过度磷酸化[55]。有研究指出,Wnt/β-catenin信号通路可以通过调控GSK-3β活性,调节Tau蛋白过度磷酸化[59]。进一步研究发现,成年转基因小鼠大脑中GSK-3β过度磷酸化导致β-catenin的核含量降低,增加了Tau蛋白磷酸化[60]。另有实验发现,ANDRO激活Wnt信号可改善年轻和APP-PS1小鼠的Tau蛋白过度磷酸化[61]。进一步研究发现,ANDRO激活Wnt信号,降低了Tau蛋白位点Thr231、Ser235以及Ser202和Thr205(AT8表位)的磷酸化水平[62]。WASP-1可激活APP-PS1小鼠Wnt信号通路,抑制Tau蛋白PHF-1(Ser396和Ser404)位点磷酸化[63]。提示激活Wnt信号通路可通过抑制GSK-3β过度磷酸化,降低其活性,改善Tau蛋白过度磷酸化。
3.3 Wnt与神经炎症
正常情况下,Wnt/β-catenin信号通路在炎症诱导的免疫应答中发挥作用,其改善神经炎症的机制是通过与其他通路的交叉作用来实现的。β-catenin是控制抗炎基因表达的激活物,过氧化物酶体增殖物激活受体γ(Peroxisome Proliferator-Activated Receptor Gamma,PPARγ)作为β-catenin的靶基因可以通过降低GSK-3β活性,激活Wnt信号通路,发挥炎症调节作用[64]。GSK-3β同时参与调节NF-κB信号通路,参与神经炎症发生。霍江涛等[65]发现川芎嗪激活AD大鼠脑内Wnt信号通路改善了大鼠脑内神经炎症。此外,大麻二酚也是通过抑制GSK-3β活性,激活Wnt信号通路,缓解神经炎症[66]。以上研究提示,抑制GSK-3β,可以激活Wnt信号通路,抑制NF-κB信号通路,从而改善神经炎症。
3.4 Wnt与自噬
大量研究表明,激活Wnt可使机体内细胞自噬水平发生变化。Wnt3a配体促进了大鼠海马组织CA1区自噬小泡的形成[67]。另有实验发现,脑外伤小鼠鼻内注射Wnt3a后,提高了小鼠大脑内Wnt3a和β-catenin表达水平,下调了自噬蛋白Beclin-1和LC3-II表达,并改善了颅脑损伤小鼠运动能力[68]。脑外伤(TBI)损害机体的认知和记忆能力,药物桑色素治疗TBI大鼠模型后,增强了Wnt-1和自噬相关标记物(LC3B II/I和Beclin-1)蛋白表达,改善了小鼠学习记忆能力[69]。目前尚未有文献直接说明AD中Wnt信号通路与自噬之间的关系,但根据以上实验推测,在AD模型中,激活Wnt信号通路可能参与调节AD中自噬活动,使得机体自噬水平正常化。
综上,激活Wnt信号通路,可以抑制Aβ产生、改善Tau蛋白过度磷酸化、减轻神经炎症,并且可使机体自噬水平正常化,改善AD认知功能障碍。
3.5 运动激活Wnt通路改善AD症状
12周跑台运动增加了STZ诱导认知障碍小鼠海马组织中Wnt-3表达,降低了GSK-3β表达,改善了小鼠认知功能障碍[70]。8周跑台运动提高了大鼠大脑皮质和海马组织中Wnt蛋白含量,增加了β-catenin稳定性,提高了Wnt受体蛋白LRP5/6含量,降低了Axin1和CK1的mRNA和蛋白水平,抑制了GSK-3β活性,说明运动激活Wnt信号通路,减少了β-catenin降解,有利于β-catenin进入细胞核完成转录,从而提高大脑学习和记忆能力[71]。另有研究亦发现,自主跑轮运动同样可以激活Wnt信号通路,增加海马组织DG区神经营养因子(Brain-Derived Neurotrophic Factor,BDNF)以及胰岛素样生长因子-1(Insulin-Like Growth Factor-1,IGF-1)表达,促进了海马组织DG区神经发生[72]。进一步研究发现,8周的认知训练增加了Tg2576大鼠海马组织中β-catenin水平,降低了Tau蛋白的过度磷酸化。以上研究表明,运动可通过激活Wnt信号通路,增加β-catenin水平,抑制GSK-3β,降低Tau蛋白过磷酸化,改善AD认知功能障碍。
4 小结与展望
综上研究,3条胰岛素信号通路与AD的发病机制密切相关。运动可通过胰岛素信号通路改善AD病理:运动可激活PI3K/AKT信号通路,抑制下游GSK-3β、GSK-3α、NF-κB、mTOR等分子,减少Aβ沉积,改善Tau蛋白过度磷酸化和神经炎症以及增加自噬水平,缓解AD病症;运动通过抑制p38MAPK和JNK信号通路,激活ERK信号通路,可降低Aβ沉积等症状;运动还可激活Wnt信号通路,增加β-catenin水平,从而改善AD认知功能障碍(图1)。
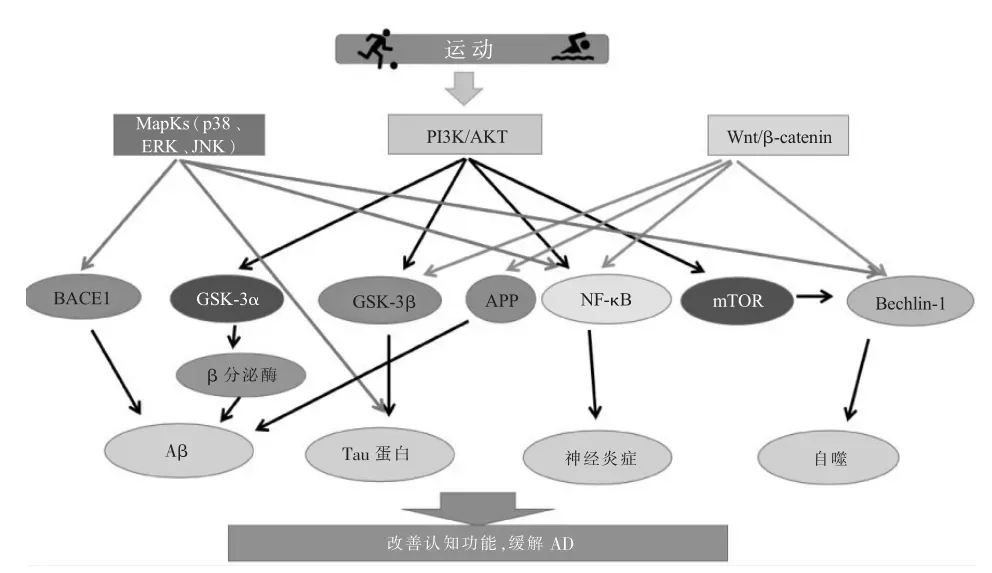
图1 胰岛素信号通路在运动改善阿尔茨海默病可能机制图Figure1 Possible mechanism of insulin signaling pathway in the improvement of Alzheimer's disease by exercise
胰岛素信号通路可能是运动改善AD的关键通路,但目前运动对AD中胰岛素信号通路调节的研究还不够全面,尤其是AD中MAPK及Wnt信号通路上下游分子的研究较少。此外,运动时间、运动强度、运动方式等因素对胰岛素信号通路的影响都需进一步深入研究。未来可进一步明晰运动对AD中胰岛素信号通路的调控作用和变化机制,深入探讨改善AD的最佳运动模式,以期为运动防治AD提供更多理论数据。
