Passivation and dissociation of Pb-type defects at a-SiO2/Si interface∗
2021-09-28XueHuaLiu刘雪华WeiFengXie谢伟锋YangLiu刘杨andXuZuo左旭
Xue-Hua Liu(刘雪华),Wei-Feng Xie(谢伟锋),Yang Liu(刘杨),and Xu Zuo(左旭),4,5,†
1College of Electronic Information and Optical Engineering,Nankai University,Tianjin 300350,China
2Microsystem and Terahertz Research Center,China Academy of Engineering Physics,Chengdu 610200,China
3Institute of Electronic Engineering,China Academy of Engineering Physics,Mianyang 621999,China
4Key Laboratory of Photoelectronic Thin Film Devices and Technology of Tianjin,Nankai University,Tianjin 300350,China
5Engineering Research Center of Thin Film Optoelectronics Technology,Ministry of Education,Nankai University,Tianjin 300350,China
Keywords:first-principles calculation,a-SiO2/Si interface,Pb-type defects,equilibrium density
1.Introduction
With the development of metal-oxide-semiconductor(MOS)technology,a-SiO2/Si interface has been widely studied as an important part of semiconductor devices.The threefold-coordinated silicon at a-SiO2/Si interface is usually called Pb-type defect,that is,there is a Si dangling bond on the side near the Si atom.The hanging bond defects at the interface will affect the performance of the device,which should be minimized in the process of semiconductor device preparation.Hydrogen is usually used to passivate Pb-type defects and inactivate them by forming the saturated dangling bond(PbH).[1]All Pb-type defects have the similar structure[·Si(−Si)3]and chemical properties.There are three types of Pbdefects:Pb,Pb0,and Pb1.The first one exists in a-SiO2/Si(111)interface,and the latter two are located at a-SiO2/Si(100)interface.[2–6]
In the field of defect physics,the microscopic recognition of charge traps at SiO2/Si(111)interface is implemented by electron paramagnetic resonance(EPR).[3,7–9]Previous works studied the passivation and dissociation kinetics of Pbdefects at SiO2/Si(111)interface by EPR.The kinetic parameters of the thermal passivation in molecular H2and dissociation in vacuum of Pbdefects were measured.[10–13]No matter passivation or dissociation,it is found that the reaction processes of Pb-type defects are very similar.[14,15]The overall analysis of Pb–hydrogen interaction in SiO2/Si(111)interface is also applicable to the defects in SiO2/Si(100)interface.[16]Therefore,Stathis and Stesmans et al.measured the kinetic parameters of Pb0and Pb1defects in the dissociation and passivation reaction processes by EPR.[15,17–19]However,due to the overlapping of the Pb0and Pb1signals,the activation energy and the pre-exponential factor of the two kinds of defects may not be accurately determined.
For the process of passivation and dissociation of Pb-type defects,the accuracy of the kinetic parameters is affected by some inevitable factors such as spectral interference.In this work,in order to accurately determine the activation energy and pre-exponential factor,the mechanism of passivation and dissociation of Pb-type defects is studied by using the Vienna ab-initio simulation package(VASP)code based on the density functional theory(DFT).The reaction rates are calculated by the climbing image nudged elastic band(CI-NEB)method and harmonic transition state theory(HTST).Then,the equilibrium density ratios of the saturated to the unsaturated ones as functions of temperature are calculated by using the reaction kinetic theory coupling the passivation and the dissociation reactions.
2.Computational methods
The a-SiO2/Si(100)interface and a-SiO2/Si(111)interface models in this work are derived from previous works.[20–22]They are prepared by molecular dynamics simulation and implemented by the LAMMPS(large scale atomic/molecular massively parallel simulator)code including the ReaxFF force field.
In order to find the reaction curve and activation energy in the passivation and dissociation processes of Pb-type defects,the climbing image-nudged elastic band(CI-NEB)method[23]is hired to search the minimum-energy path(MEP)and exact saddle point,and transition state structure.Firstly,stable initial and final structures are prepared,and then appropriate number of images are inserted between the initial and final structures.The Perdew–Burke–Ernzerhof(PBE)[24]parameterization of the generalized gradient approximation(GGA)exchange correlation is hired in the calculation.The interaction between valence electrons and atomic core are described in terms of the projection augmented plane wave method.The cutoff energy is set to be 520 eV for the plane-wave basis.The precision of electron iteration is 10−5eV,and the precision of ionic loop is controlled by the criterion that the residue force on each atom is less than 0.05 eV/˚A.
After the transition state is found,the inactive atoms in the initial state and the transition state are fixed,and the harmonic vibration frequency is calculated.Then,the reaction rate is calculated in terms of the harmonic transition state theory(HTST).[25]
3.Kinetics of passivation and dissociation
Brower quantified the interaction kinetics of Pband H2at SiO2/Si(111)interface in his early work.The essence of the interaction is the dissociation of H2molecule,and Pbacts as a catalyst for the decomposition of H2molecule.It is discovered that when temperature is higher than approximately 220°C,Pbcenters are passivated to PbH in H2.In addition,when temperature exceeds 550°C,the passivated centers dissociate in vacuum.[10,14]The passivation and dissociation are described by the equations
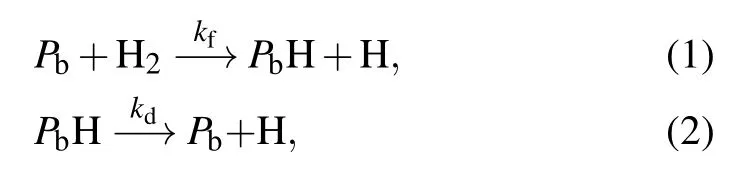
where kfis the rate constant of the passivation and kdis the rate constant of the dissociation.Because H atoms enter vacuum through SiO2,they have a very high diffusion rate and leave from the interface almost instantaneously.As a result,reverse reactions can be ignored.[16]The reaction rate equations of the passivation and dissociation are as follows:
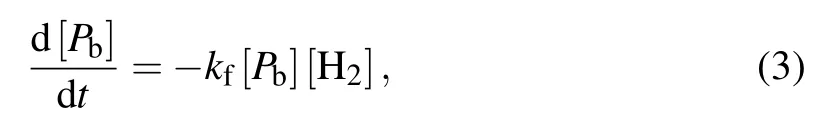
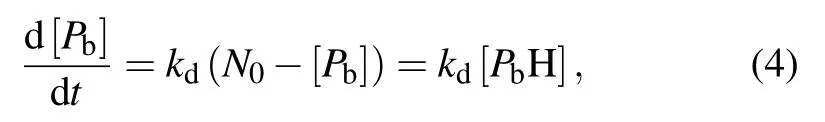
where N0is the total density of Pbsites(the maximum number of recoverable ESR active defects),[26]N0~5×1012cm−2,and[H2]is the volume concentration of H2molecules at SiO2/Si interface.By combining Eqs.(3)and(4),the equilibrium density ratio of[PbH]to[Pb]is obtained by

where[H2]corresponds to the physical solubility of H2in vitreous silica,so for the purpose of calculation,it can be approximated by the stretched exponential[16,27]
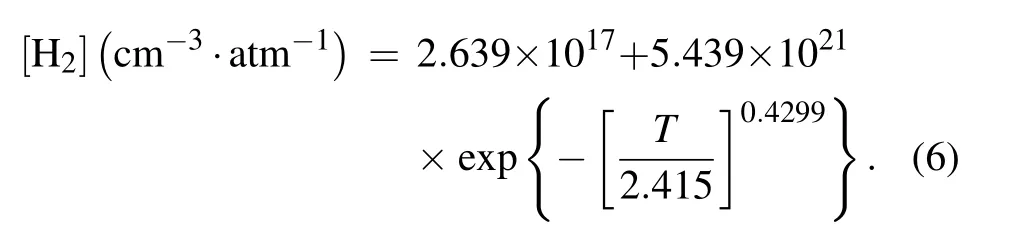
Therefore,in order to obtain the equilibrium density ratios of[PbH]to[Pb]as functions of temperature and hydrogen concentration,we only need to know the passivation reaction rate constant kfand the dissociation reaction rate constant kd.The passivation and dissociation rate constants are given by the Arrhenius equations[11]

where T is the temperature(in units of K),Efis the activation energy of the passivation reaction,Edis the activation energy of the dissociation reaction,kBis the Boltzmann’s constant,and kf0and kd0are the respective pre-exponential factors for passivation and dissociation.
According to the harmonic transition state theory(HTST),the pre-exponential factor is given by
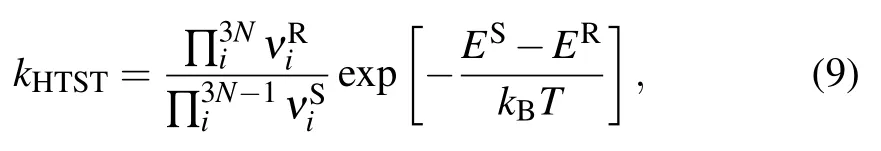
Pb0and Pb1defects are located at SiO2/Si(100)interface.Like Pbdefect,their passivation and dissociation reactions mainly take place under hydrogen annealing,where the above analysis holds.[14–16]The initial density[28]of Pb0and Pb1defects is about 1×1012cm−2,and the initial density of Pbdefects is about 5×1012cm−2.
4.Results and discussion
4.1.Kinetic parameters of Pb-type defects
4.1.1.Passivation of Pb defects with molecular H2
The Pbdefect is located at SiO2/Si(111)interface and has a certain effect on the surface states of the semiconductoroxide interface.[2,29]In the process of annealing,[30]the interface defects will be passivated in H2.First of all,we calculate the reaction barrier and look for the transition state of the passivation reaction by using the CI-NEB method.The structures of the initial and final states of the passivation reaction are shown in Fig.1(a).In order to obtain the initial state of the passivation reaction,a hydrogen molecule is placed in the position near the trivalent interfacial silicon atom,and then the atomistic model is relaxed.In order to obtain the final state of the passivation reaction,one hydrogen atom is fixed in the interstitial position near the passivated Pbdefect,and the other hydrogen atom is used to passivate the silicon dangling bond.
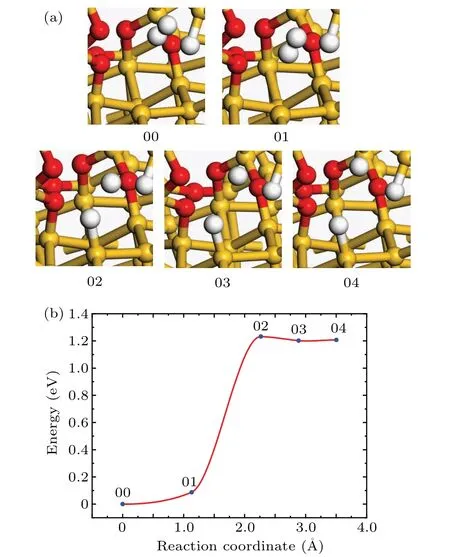
Fig.1.(a)The atomic structures of the initial(00),final(04),and intermediate(01,02,and 03)states of the Pb passivation reaction.(b)Reaction curve of the Pb passivation reaction between H2 and Pb defect.
At the beginning of the reaction,the bond length of the H–H bond in hydrogen molecule is about 0.75˚A.With the reaction proceeding,the distance between the two H atoms increases,and the total energy of the system also increases.When the reaction proceeds to the 02 state,the distance between the two hydrogen atoms is about 1.76˚A,and the distance between the passivating hydrogen atom and the defective silicon atom(Sid)is 1.52˚A.At this time,the energy reaches the maximum,and the energy increases by 1.23 eV compared with the initial state.With the reaction going on,when the final state is reached,the H and Siddevelop a bond with a bond length of 1.5˚A,which is consistent with the ideal Si–H bond length(~1.5˚A).From the transition state 02 to the final state 04,the total energy of the system decreases by 0.025 eV.
In Fig.1(b),the 02 state is the transition state and the activation energy is 1.23 eV.The vibrational frequency calculated by VASP software shows that there is no imaginary frequency in the initial state and one and only one imaginary frequency in the transition state.The pre-exponential factor is calculated to be 1.54×10−9cm3·s−1.
4.1.2.Dissociation of PbH in vaccum
In the process of annealing in hydrogen-containing ambient,both passivation and dissociation of Pbdefects occur.[16]The CI-NEB method is hired to calculate the reaction barrier of the dissociation reaction and to look for the transition state.The structures of the initial and final states of the dissociation reaction are shown in Fig.2(a).In order to obtain the initial state of the dissociation reaction,a hydrogen atom is moved to the unsaturated dangling bond to passivate the Pbdefect,and then the whole model is relaxed.In order to obtain the final state of the dissociation reaction,the hydrogen atom is placed in the void of the a-SiO2random network far enough away from the Pbdefect,and then the whole model is relaxed.
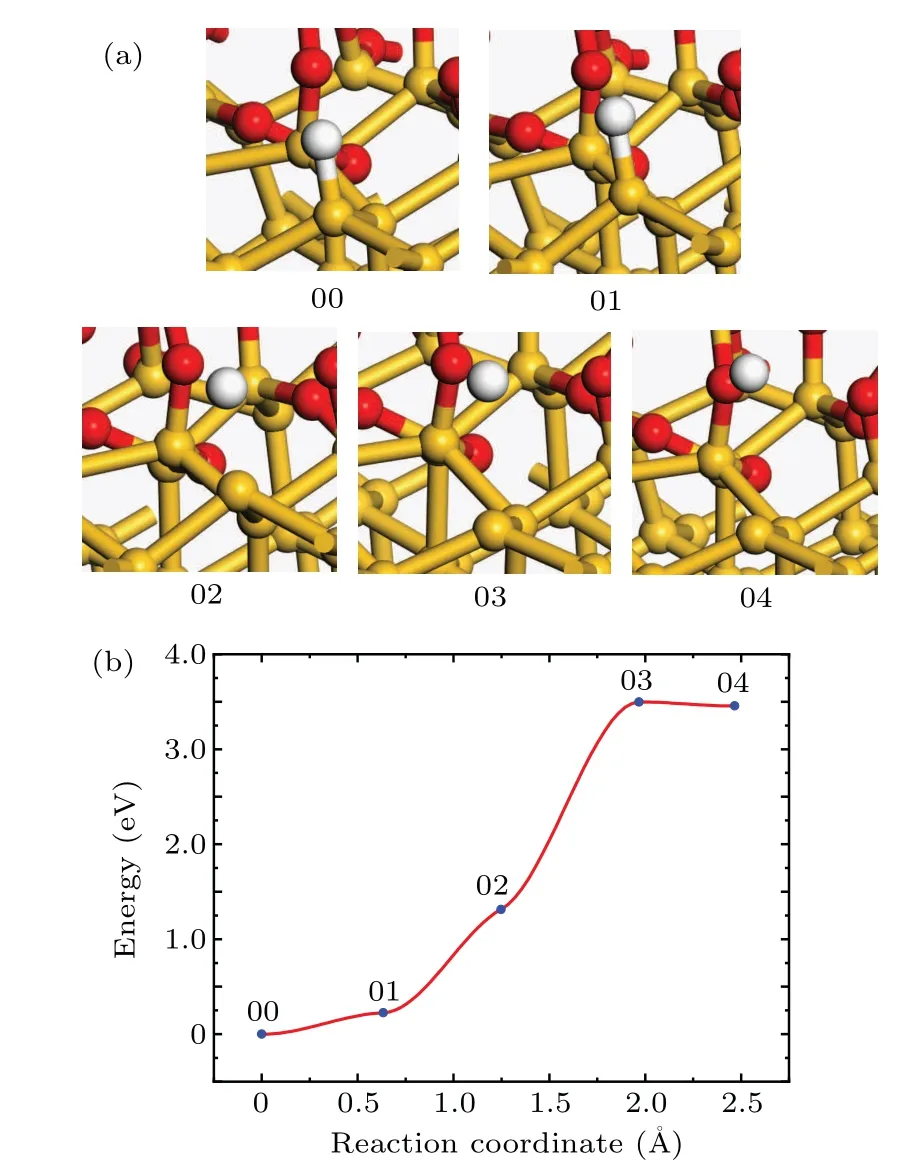
Fig.2.(a)The atomic structures of the initial(00),final(04),and intermediate(01,02,and 03)states of the Pb dissociation reaction.(b)Reaction curve of the Pb dissociation reaction between H atom and Pb defect.
At the beginning of the dissociation reaction,the Si–H bond length is 1.5˚A.With the reaction proceeding,the hydrogen atom leaves from the Sid.With the reaction going on,the distance between the hydrogen atom and the Sidbecomes longer and the energy of the system increases.When the distance between the hydrogen atom and the Sidis about 2.95˚A,the total energy of the system reaches the maximum about 3.5 eV(transition state 03 in Fig.2(b))higher than the initial state.As the reaction proceeds to the final state,the distance between the hydrogen atom and the Sidis about 3.35˚A,and the total energy of the system decreases by 0.04 eV.
In Fig.2(b),the 03 state is the transition state and the activation energy is 3.50 eV.The frequency analysis of simple harmonic vibration shows that there is no imaginary frequency in the initial state,and there is one and only one imaginary frequency in the transition state.The pre-exponential factor is 4.12×1014s−1.
Both passivation and dissociation simulated here are strongly endothermic,because a neutral hydrogen atom is produced in the interstitial of the a-SiO2random network.
4.1.3.Passivation of Pb1 defects with molecular H2
For Pb1defects,Stirling et al.pointed out that the asymmetrically oxidized dimer(AOD)model is more reasonable than the dimer and bridge model.[31]In the AOD model,two backward Si–Si bonds in the dimer structure are oxidized,which makes the direction of the dangling bond closer to the experimental value.Therefore,the passivation of the AOD model is investigated in this work.The structures of the initial and final states of the Pb1passivation reaction are shown in Fig.3(a).In the passivation process,the passivating hydrogen atom is detached from the hydrogen molecule and approaching to the Pb1defect.Due to the interaction between the passivating hydrogen atom and the dangling band Si,the distance between the two hydrogen atoms is increasing,and the total energy of the system increases.When the distance between the two hydrogen atoms is about 1.41˚A and the distance between passivating hydrogen atom and Sidis about 1.63˚A,the energy increases by 1.19 eV,and the total energy of the system reaches the maximum value(transition state 02 in Fig.3(b)).As the reaction continues,the distance between passivating hydrogen atom and Sidbecomes shorter,and the total energy of the system decreases by 0.077 eV.
In Fig.3(b),the state 02 is the transition state and the activation energy is 1.19 eV.The frequency analysis of simple harmonic vibration indicates that there is no imaginary frequency in the initial state and one and only one imaginary frequency in the transition state.The pre-exponential factor is 1.49×10−9cm3·s−1.
4.1.4.Dissociation of Pb1H in vacuum
The structures of the initial and final states of the Pb1dissociation reaction are shown in Fig.4(a).In the process of dissociation reaction,the hydrogen atom will be detached from the passivated Si atom,and the distance between the hydrogen atom and the Sidbecomes longer.In addition,the total energy of the system increases slightly.When the distance between the hydrogen atom and the Sidis 2.99˚A,the Sid–H bond is broken,and the total energy of the system reaches the maximum value,which is about 3.58 eV(transition state 03 in Fig.4(b)).With the reaction going on,the total energy of the system decreases by 0.026 eV,and the distance between hydrogen atom and Sidreaches about 3.39˚A.
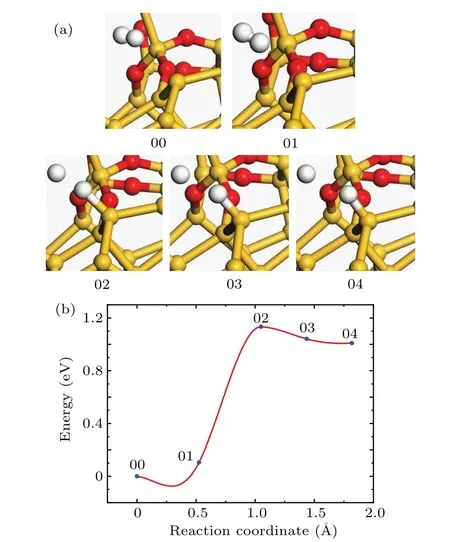
Fig.3.(a)The atomic structures of the initial(00),final(04),and intermediate states(01,02,and 03)of the Pb1 passivation reaction.(b)Reaction curve of the Pb1 passivation reaction between H2 and Pb1 defect.

Fig.4.(a)The atomic structures of the initial(00),final(04),and intermediate(01,02,and 03)states of the Pb1 dissociation reaction.(b)Reaction curve of the Pb1 dissociation reaction between H atom and Pb1 defect.
In Fig.4(b),the 03 state is the transition state and the activation energy is 3.58 eV.The results of frequency calculation demonstrate that there is no imaginary frequency in the initial state and one and only one imaginary frequency in the transition state.The pre-exponential factor is 2.33×1014s−1.
4.1.5.Passivation of Pb0 defects with molecular H2
In addition to Pb1defects,there are Pb0defects in a-SiO2/Si(100)interface.The Pb0defect is located in the second layer of silicon atom beneath the interface.[32]The structures of the initial and final states of the Pb0passivation reaction are shown in Fig.5(a).In the passivation process,the passivating hydrogen atom is detached from the hydrogen molecule and approaching to the Pb0defect.Due to the Coulomb interaction between the hydrogen atom and the Sid,the distance between the hydrogen atom and Sidis getting closer,the distance between two hydrogen atoms is increasing,and the total energy of the system is rising.Subsequently,the H–H bond is broken and the hydrogen atom near the defect is covalently bonded to the Sid(transition state 02 in Fig.5(b)).Meanwhile,the distance between the two hydrogen atoms is about 1.40˚A,and the distance between the passivating hydrogen atom and the Sidis 1.72˚A.With respective to the initial state,the total energy of the system increases by 1.13 eV.With the reaction going on,the distance between the hydrogen atom and the Sidgradually becomes shorter,and the total energy of the system decreases by 0.123 eV.
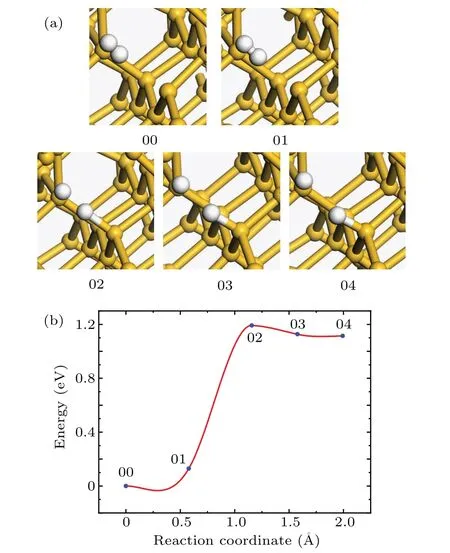
Fig.5.(a)The atomic structures of the initial(00),final(04),and intermediate(01,02,and 03)states of the Pb0 passivation reaction.(b)Reaction curve of the Pb0 passivation reaction between H2 and Pb0 defect.
In Fig.5(b),the state 02 is the transition state and the activation energy is 1.13 eV.The results of frequency analysis display that there is no imaginary frequency in the initial state and one and only one imaginary frequency in the transition state.The pre-exponential factor is 2.49×10−8cm3·s−1.
4.1.6.Dissociation of Pb0H in vaccum
Compared to Pb1defect,the dissociation of Pb0defect is more complicated.Because the atomic structure around a Pb0defect is similar to that of crystalline silicon,the dissociated hydrogen atom is easily trapped by a Si–Si bond during the dissociation process,forming the stable hydrogen bridge structure(Si–H–Si).The structures of the initial and final states of the dissociation reaction are shown in Fig.6(a).A hydrogen atom is transferred to the unsaturated dangling bond to passivate the Pb0defect.After structural relaxation,the initial state structure of the Pb0defect dissociation reaction is obtained.The hydrogen atom is placed next to the Si–Si bond near the defect,and the final state structure of Pb0defect dissociation reaction is obtained by structural relaxation.
In the process of dissociation,the hydrogen atom is separated from the Sid.As the reaction continues,the distance between the hydrogen atom and the Sidbecomes longer and the total energy of the system increases.In the 03 state,the covalent bond between the hydrogen atom and the Sidis broken.In addition,the hydrogen atom is bonded with Si(1),and the length of Si(1)–H bond is about 1.76˚A.In the 04 state,the distance between the hydrogen atom and the Sidis about 2.67˚A,and the Si(1)–H bond length is about 1.58˚A.The energy of the system reaches the maximum value,which increases by about 2.25 eV.In the 05 and 06 states,the atomic structures around Si(1)and Si(2)continue to relax.When the distance between the hydrogen atom and the Sidreaches about 3.75˚A,the hydrogen atom is bonded with both Si(1)and Si(2)forming the hydrogen bridge structure,and the total energy of the system decreases by 0.295 eV.The Si(1)–H bond length is about 1.61˚A,and the Si(2)–H bond length is about 1.65˚A.
In Fig.6(b),the 04 state is the transition state and the activation energy is 2.25 eV.The results of the frequency analysis show that there is no imaginary frequency in the initial state and one and only one imaginary frequency in the transition state.The pre-exponential factor is 5.99×1013s−1.
Compared to Pband Pb1defects,the forward reaction barrier of Pb0defect is obviously lower.Through Bader charge analysis of the dissociation process of Pb,Pb0,and Pb1defects,it is found that the initial states of the dissociation reactions for the three defects are basically similar:the valence electron numbers of the three passivated Si atoms are 3.34,3.39,and 3.15,respectively.The valence electron number of the passivating hydrogen atoms of the three defects are 1.55,1.59,and 1.55,respectively.For the three defects,the final states of the dissociation reactions are slightly different:the valence electron numbers of the three defects are 3.98,3.87,and 3.91,respectively.The valence electron numbers of the passivating hydrogen atoms of the three defects are 0.99,1.64,and 0.99,respectively.Therefore,the charge transfer of Pb0dissociation is very different from that of Pband Pb1.The final state of the Pb0dissociation produces the more stable hydrogen bridge structure,which makes the hydrogen atom negatively charged.This may be the reason why the forward reaction barrier of the Pb0dissociation reaction is lower.
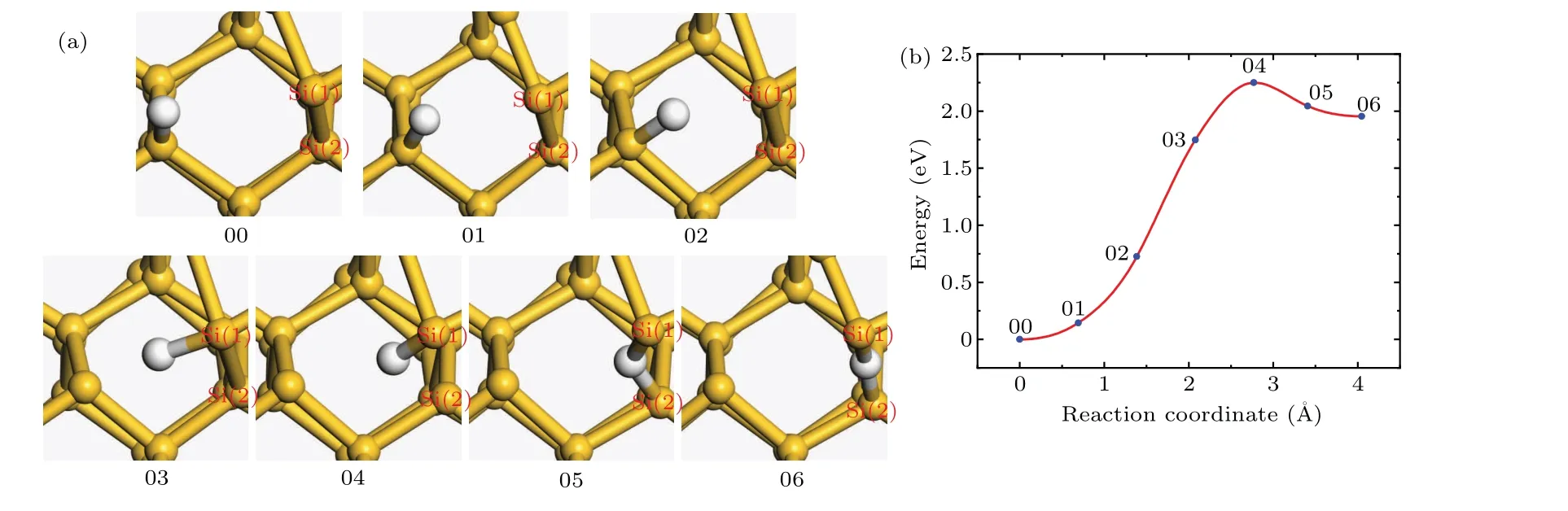
Fig.6.(a)The atomic structures of the initial(00),final(06),and intermediate(01,02,03,04,and 05)states of the Pb0 dissociation reaction.(b)Reaction curve of the Pb0 dissociation reaction between H atom and Pb0 defect.
4.2.Equilibrium density ratio of saturated interface dangling bond and intebrface defect
The calculated values of this work are compared with the experimental values in Tables 1 and 2.The calculated results of this work agree well with the experimental values.σEfand σEdare spreads in activation energies around the mean values Efand Edfor passivation and dissociation,respectively.The spreads are found adequately described by Gaussian distributions in the generalized simple thermal(GST)model.[16]In the passivation reactions of the three kinds of defects,the activation energies are approximately equal and Pb>Pb1>Pb0.In the dissociation reactions,the activation energies of Pband Pb1are approximately equal,but the activation energy of the Pb0defect is lower than them by about 1 eV.Because the hydrogen atom generated by the Pb0dissociation reaction is not standing alone in bulk silicon and forms the stable hydrogen bridge(Si–H–Si)structure,the activation energy of the dissociation reaction is reduced.In addition,the pre-exponential factors kf0of the passivation reactions of the three kinds of defects are relatively close to each other and Pb0>Pb>Pb1,in which the pre-exponential factor of Pb0passivation reaction is one order of magnitude larger than that of Pband Pb1.The preexponential factors kd0of dissociation reactions are also close to each other and Pb>Pb1>Pb0,where the pre-exponential factor of Pb0dissociation reaction is one order of magnitude smaller than that of Pband Pb1.This result may be determined by the structure of the defect itself.It is worth noting that the pre-exponential factors of the passivation reactions of Pb0and Pb1calculated in this work are slightly lower than the experimental values,possibly because of the fact that the signals of Pb0and Pb1defects overlap each other,resulting in spectral interference.[19]
We set the temperature range of 600–1000°C with an interval of 20°C,and substitute it into Eq.(6)to obtain the solubility of H2in SiO2at different temperatures.The calculated activation energies and pre-exponential factors of the passivation and dissociation reactions of the Pb-type defects are substituted into Eqs.(7)and(8)to obtain the reaction rate constants kfand kdat different temperatures.By substituting kf,kd,and[H2]into Eq.(5),the equilibrium density ratio of the saturated interfacial dangling bonds and interface defects under the impact of both passivation and dissociation is obtained,as shown in Fig.7.
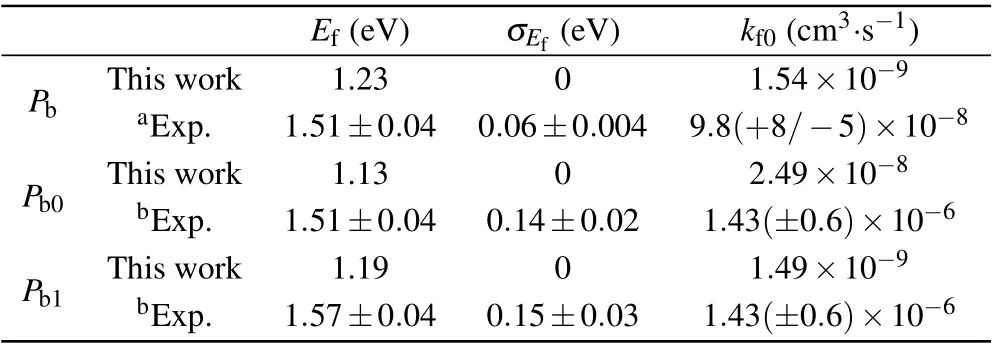
Table 1.Summary of the kinetic parameters for passivation reactions of Pb-type defects.
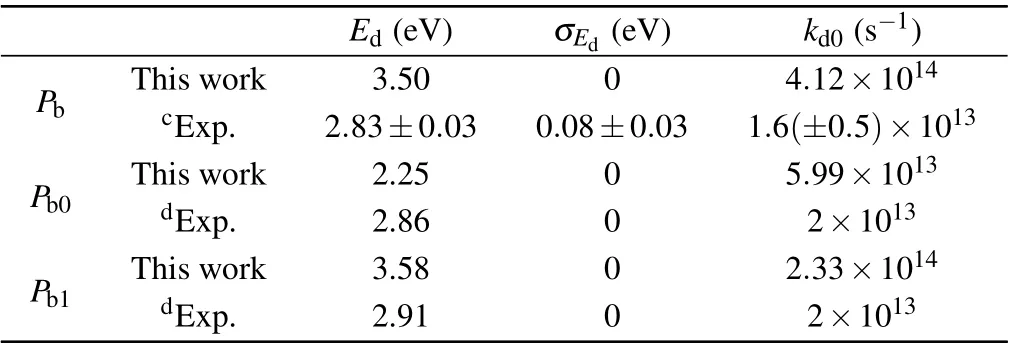
Table 2.Summary of the kinetic parameters for dissociation reactions of Pb-type defects.
It is shown in Fig.7 that the equilibrium density ratio of the saturated interfacial dangling bonds to the interfacial defects monotonically decreases with the increase of temperature in the range of 600–1000°C.It is consistent with Stesman’s view that more effective passivation requires longer passivation at lower temperatures.[16]Among the Pb-type defects,the value of[Pb1H]/[Pb1]is always the largest,the value of[PbH]/[Pb]is the second,and the value of[Pb0H]/[Pb0]is the smallest.At 600°C,the maximum of[Pb1H]/[Pb1]is about 1.09×108,that of[PbH]/[Pb]is about 1.29×107,and that of[Pb0H]/[Pb0]is about 3.36×102.At 1000°C,the value of[Pb1H]/[Pb1]is about 4.78×103,that of[PbH]/[Pb]is about 9.37×102,and that of[Pb0H]/[Pb0]is about 2.96.This is because the activation energy of Pb0dissociation reaction is much lower than those of Pband Pb1.In Arrhenius formula,the activation energy is in the exponential position,and a relatively low activation energy will lead to a significant increase in the rate constant of the dissociation reaction.It is obvious in Eq.(5)that the[Pb0H]/[Pb0]is inversely proportional to the dissociation rate constant kd.This result is consistent with that of Stathis et al.[33,34]Because Pb0defect is located in the second atomic layer beneath the transition region,Pb0defect is more difficult to be passivated than Pb1defect.
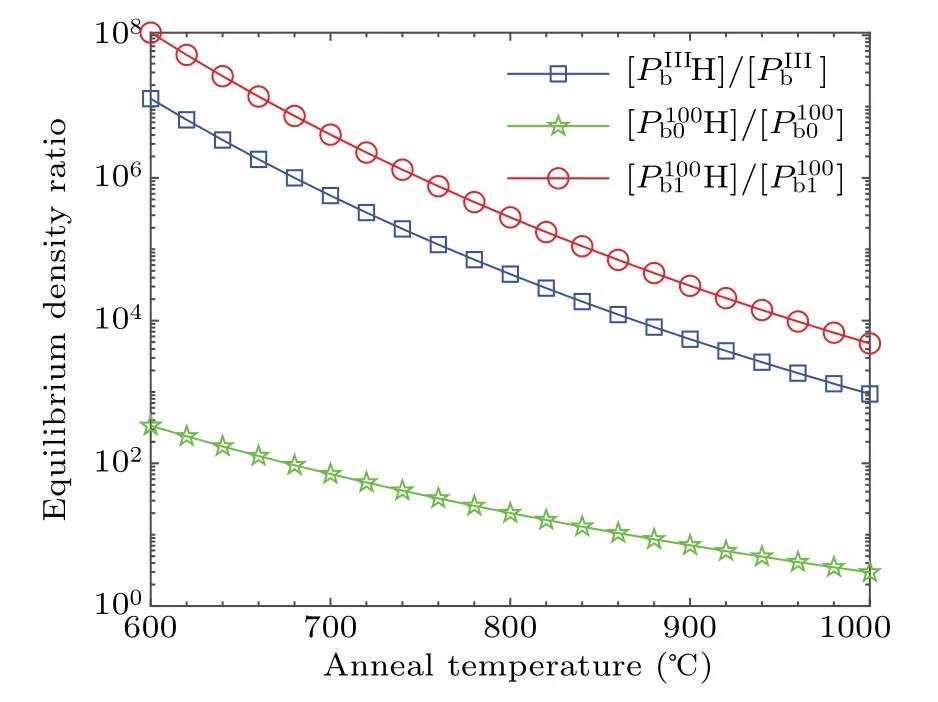
Fig.7.Equilibrium density ratio of saturated interfacial dangling bonds and interfacial defects of Pb-type defects at 1 atm H2.
5.Conclusion
Firstly,based on the kinetic theory of Pb–hydrogen reactions,it is deduced that the ratio of equilibrium density of the saturated interfacial dangling bonds to interfacial defects is a function(Eq.(5))of reaction rate constants kfand kd.Secondly,the reaction mechanisms of passivation and dissociation of the Pb-type defects are investigated by using first-principles calculations based on density functional theory.The activation energies and transition states of the passivation and dissociation reactions of Pb-type defects are calculated by the CI-NEB method.According to the harmonic transition state theory(HTST),the harmonic vibration frequency of the initial state and transition state is analyzed,and the pre-exponential factor is calculated.The calculated results are essentially consistent with the previous experimental values.Finally,the calculated activation energy and pre-exponential factor are substituted into the Arrhenius formula to obtain the reaction rate constant,and the reaction rate constant is substituted into Eq.(5)to obtain the equilibrium density ratio of the saturated interfacial dangling bonds and interfacial defects.
In the passivation reactions,the passivating hydrogen atom is detached from the hydrogen molecule and approaching to the interface defect to passivate the dangling bond defect.In the dissociation reactions,the hydrogen atom breaks away from the three-fold coordinated silicon atom and reaches the void near the interface defects.However,the hydrogen atom in the Pb0dissociation reaction can be easily trapped by a Si–Si bond,forming the Si–H–Si hydrogen bridge structure.The simulation results show that the passivation and dissociation reactions of Pb-type defects are endothermic,and the forward reaction barrier is much higher than the reverse one.
In brief,the activation energies and pre-exponential factors of the passivation and dissociation reactions of the Pbtype defects are calculated by using first-principles calculations and in good agreement with the experimental values.It is shown that the ratio of equilibrium density of the saturated interfacial dangling bonds to interfacial defects follows the order Pb1>Pb>Pb0.When the temperature is between 600°C and 1000°C,the passivation degree of Pbtype defects monotonically decreases with the increase of temperature.Among them,the passivation degree of Pb0is the lowest.
杂志排行

Chinese Physics B的其它文章
- Multiple solutions and hysteresis in the flows driven by surface with antisymmetric velocity profile∗
- Magnetization relaxation of uniaxial anisotropic ferromagnetic particles with linear reaction dynamics driven by DC/AC magnetic field∗
- Influences of spin–orbit interaction on quantum speed limit and entanglement of spin qubits in coupled quantum dots
- Quantum multicast schemes of different quantum states via non-maximally entangled channels with multiparty involvement∗
- Magnetic and electronic properties of two-dimensional metal-organic frameworks TM3(C2NH)12*
- Preparation of a two-state mixture of ultracold fermionic atoms with balanced population subject to the unstable magnetic field∗