Fe(Ⅲ)强化单宁酸原位修复Cr(Ⅵ)污染含水层反应机理及效能
2021-09-22滑钰铎秦雪铭杨新如赵勇胜
滑钰铎, 秦雪铭, 杨新如, 赵勇胜
(1. 吉林大学 新能源与环境学院, 长春 130021; 2. 吉林大学 石油化工污染场地控制与修复技术国家地方联合工程实验室, 长春 130021)
变价重金属铬(Cr)导致的土壤和地下水污染是环境污染中最严重、 最常见的重金属污染问题之一[1-2]. Cr一般以三价和六价两种形式存在, 其中Cr(Ⅵ)具有较强的迁移能力和毒性[3-4]. 目前, 尽管各行业已开始严格控制Cr(Ⅵ)排放, 但土壤和地下水中仍存在不同程度的Cr(Ⅵ)污染, 严重威胁生态系统和人类的安全[5-10]. 因此, 寻找高效、 环保的Cr(Ⅵ)污染修复方法至关重要.
目前, 常用的化学还原修复Cr(Ⅵ)地下水污染的方法是用各种还原剂将Cr(Ⅵ)还原为Cr(Ⅲ), 然后通过调节pH值使Cr(Ⅲ)沉淀, 达到去除总铬的目的[11-13]. 但传统修复工艺在使用过程中存在一定的缺陷: 在去除总铬时引入外源碱性物质增加了修复成本; 一些还原剂在土壤中不能降解, 进而对地下环境产生影响.
单宁酸(TA)比传统的化学还原剂更环保, 可作为电子供体, 并在一定程度上改善土壤微生物群落和功能[14]. 但单独使用TA时对Cr(Ⅵ)的去除效率较低, 达不到应用要求. TA吸附Cr(Ⅲ)后, 由于环境条件发生变化, Cr(Ⅲ)易发生解吸[15], 因此, 需利用电子穿梭体提高电子转移效率, 并将总铬以一种更稳定的状态从体系中去除. Fe(Ⅲ)作为电子穿梭体可有效提高电子转移效率, 但目前对Fe(Ⅲ)强化多酚修复Cr(Ⅵ)污染含水层机理的研究结果尚不统一: Chrysochoou等[15]研究表明, 多酚还原Fe(Ⅲ)生成纳米零价铁(nZVI); Mystrioti等[16]研究表明, 多酚与Fe(Ⅲ)间的反应导致形成Fe(Ⅱ,Ⅲ)纳米颗粒; Wang等[17]研究表明, 有机物作为电子供体还原过渡变价金属, 还原产物可继续还原Cr(Ⅵ).
基于此, 本文以Fe(Ⅲ)为强化试剂对TA原位修复Cr(Ⅵ)污染含水层进行强化, 研究不同条件下强化效果的变化, 并通过多种表征方法对强化机理进行分析, 对Fe(Ⅲ)强化TA原位修复Cr(Ⅵ)污染含水层提供理论支持.
1 实 验
1.1 试剂和材料
六水合氯化铁(FeCl3·6H2O)、 单宁酸(TA)、 二苯碳酰二肼(C13H14N4O)、 重铬酸钾(K2Cr2O7)、 硫酸(H2SO4)、 盐酸(HCl)、 磷酸(H3PO4)等试剂均购自北京化工厂和国药化学试剂有限公司; 所有溶液均采用去离子水配制. 将TA, K2Cr2O7和FeCl3·6H2O溶于超纯水中制备Cr(Ⅵ)和Fe(Ⅲ)储备液, 最终质量浓度分别为9,1 g/L和160 mg/L.
1.2 实验方法
1.2.1 Fe(Ⅲ)强化TA修复Cr(Ⅵ)污染含水层
设置3组反应体系, 每组实验设置3个棕色反应瓶作为平行实验. 恒温25 ℃条件下, 先分别量取5.00 mL Cr(Ⅵ)储备液置于3组200 mL棕色反应瓶中; 再分别量取10.00 mL去离子水、 10.00 mL去离子水、 10.00 mL Fe(Ⅲ)储备液依次加入3组棕色反应瓶中; 然后用去离子水将各反应容器中反应体系增至95.00 mL; 最后在第二组与第三组反应瓶中加入5.00 mL TA储备液以触发反应.
1.2.2 TA初始质量浓度对修复Cr(Ⅵ)污染含水层的影响
设置5组反应体系, 每组实验设置3个棕色反应瓶作为平行实验. 恒温25℃条件下, 先分别量取5.00 mL Cr(Ⅵ)储备液置于5组200 mL棕色反应瓶中; 再分别量取5.00 mL Fe(Ⅲ)储备液加入棕色反应瓶中; 然后用去离子水将各反应容器中反应体系分别增至99.00,97.00,95.00,93.00,91.00 mL; 最后分别量取1.00,3.00,5.00,7.00,9.00 mL TA储备液依次加入5组反应体系以触发实验.
1.2.3 Fe(Ⅲ)初始质量浓度对修复Cr(Ⅵ)污染含水层的影响
设置5组反应体系, 每组实验设置3个棕色反应瓶作为平行实验. 恒温25 ℃条件下, 先分别量取5.00 mL Cr(Ⅵ)储备液置于5组200 mL棕色反应瓶中; 再分别量取1.00,2.00,3.00,4.00,5.00 mL Fe(Ⅲ)储备液加入棕色反应瓶中; 然后用去离子水将各反应容器中反应体系增至95.00 mL; 最后分别量取5.00 mL TA储备液依次加入5组反应体系以触发实验.
为模拟地下水环境, 反应过程中对容器加盖进行隔绝空气处理, 避免溶解氧对反应结果产生影响. 为避免生物还原对反应结果产生影响, 在实验前对上述溶液均进行灭菌处理. 反应前将所有反应体系的pH值调为4.0, 并置于25 ℃的水浴摇床中振荡. 对反应体系中的颗粒相和溶解相进行分离, 并对溶解相中Cr(Ⅵ)和Fe(Ⅲ)的质量浓度进行测定, 确定Cr(Ⅵ)还原效果及Fe(Ⅲ)质量浓度的变化.
1.3 铁形态和铬形态分析
根据上述实验结果, 选取修复Cr(Ⅵ)污染含水层的最适TA初始质量浓度及Fe(Ⅲ)初始质量浓度, 重复进行Fe(Ⅲ)强化植物多酚修复Cr(Ⅵ)污染含水层效果实验, 并于既定的时间间隔取出相同量的反应溶液. 反应溶液溶解相和颗粒相的分离操作如下: 取出5.00 mL反应溶液, 其中1.00 mL用H2SO4+H3PO4消化, 稀释后利用火焰原子吸收法和分光光度法分别测定溶液中总Cr(Crtot)和Cr(Ⅵ)的质量浓度. 剩余溶液用0.22 μm聚醚矾膜(天津金腾有限公司)过滤, 并立即分析溶解态Cr(Crdi)和Cr(Ⅵ)(Cr(Ⅵ)di)的质量浓度. 由于Cr主要以Cr(Ⅲ)和Cr(Ⅵ)形式存在, 因此颗粒相的Cr(Ⅲ)(Cr(Ⅲ)p)可通过如下公式计算:
Cr(Ⅲ)di=Crdi-Cr(Ⅵ)di,
(1)
Cr(Ⅲ)p=Crtot-Cr(Ⅵ)tot-Cr(Ⅲ)di.
(2)
为研究植物多酚/Fe(Ⅲ)体系修复Cr(Ⅵ)污染含水层的机理, 实验测定了在有无Cr(Ⅵ)存在情形下Fe的物态变化与质量浓度. Fe在不同物态下的测定方法和Cr基本相同, 总铁(Fetot)和溶解态Fe(Ⅲ)(Fe(Ⅲ)di)的质量浓度可通过分光光度法与火焰原子吸收法测定, 而溶解态Fe(Ⅱ)(Fe(Ⅱ)di)与颗粒Fe(Fep)可通过如下公式计算:
Fe(Ⅱ)di=Fedi-Fe(Ⅲ)di,
(3)
Fep=Fetot-Fedi.
(4)
1.4 分析方法
实验采用二苯碳酰二肼分光光度法在波长540 nm处测定Cr(Ⅵ)di的质量浓度, 采用硫氰酸盐比色法在485 nm处测定Fe(Ⅲ)di的质量浓度. 用分光光度计UV-2600(A)测量, 其检出限分别为4 μg/L(0.077 μmol/L)和0.03 mg/L(0.54 μmol/L).
用火焰原子吸收法测定Crtot和Fetot的质量浓度, 使用仪器为火焰原子吸收光谱仪(AA-6300型, 岛津(上海)实验器材有限公司), 检出限分别为0.1,0.03 mg/L. 每10个样品分析一次质量控制标准, 以保持与标准曲线的偏差≤5%.
1.5 表征方法
按照既定的时间间隔吸取反应溶液, 对颗粒相和溶解相进行分离, 并将Cr(Ⅵ)完全还原后的样品分别进行表征. 方法如下: 反应溶液以10 000 r/min离心10 min后, 用0.22 μm聚醚矾膜对溶液进行抽滤. 通过上述两种方法收集到的颗粒物质于55 ℃真空干燥并低温保存. 样品使用X射线光电子能谱进行分析. 结合能用284.8 eV的C1s峰进行标定, 通过RELEASE-INSTALLATION-Avantage软件完成各区域曲线的拟合. 溶解相通过扫描型紫外-可见分光光度计进行紫外光谱扫描, 以确定反应前后有机物中官能团的变化.
2 结果与讨论
2.1 Fe(Ⅲ)强化TA修复地下水Cr(Ⅵ)污染效果
图1为Fe(Ⅲ)、 TA和TA/Fe(Ⅲ)体系对Cr(Ⅵ)污染地下水的还原效率及Fe(Ⅲ)质量浓度的变化. 由图1可见: 单独加入Fe(Ⅲ)时, Cr(Ⅵ)未被还原; 单独加入TA, 到达反应终点时约有60% 的Cr(Ⅵ)被还原; 当Fe(Ⅲ)与Cr(Ⅵ)预混合并加入TA触发反应后, 体系内的Cr(Ⅵ)在5 min内约降低77%, 之后还原速率减慢, 在120 min时Cr(Ⅵ)的去除率达99.9%. 结果表明: 单独的Fe(Ⅲ)与Cr(Ⅵ)无法发生反应, 单独的TA对Cr(Ⅵ)有一定的还原效果, 但还原效率较低; 加入Fe(Ⅲ)可有效提高TA对Cr(Ⅵ)的还原速率, 并提高Cr(Ⅵ)最终的还原量. TA作为天然多酚, 在结构上具有大量的酚羟基, 因而对Fe(Ⅲ)有较强的结合能力. 在该过程中, TA上的两个邻位酚羟基以氧负离子的形式与作为中心离子的Fe(Ⅲ)形成了稳定的环状结构[15,18-19]. 而Fe(Ⅲ)作为良好的电子穿梭体, 为电子由酚羟基传递到Cr(Ⅵ)上提供了便捷的途径.
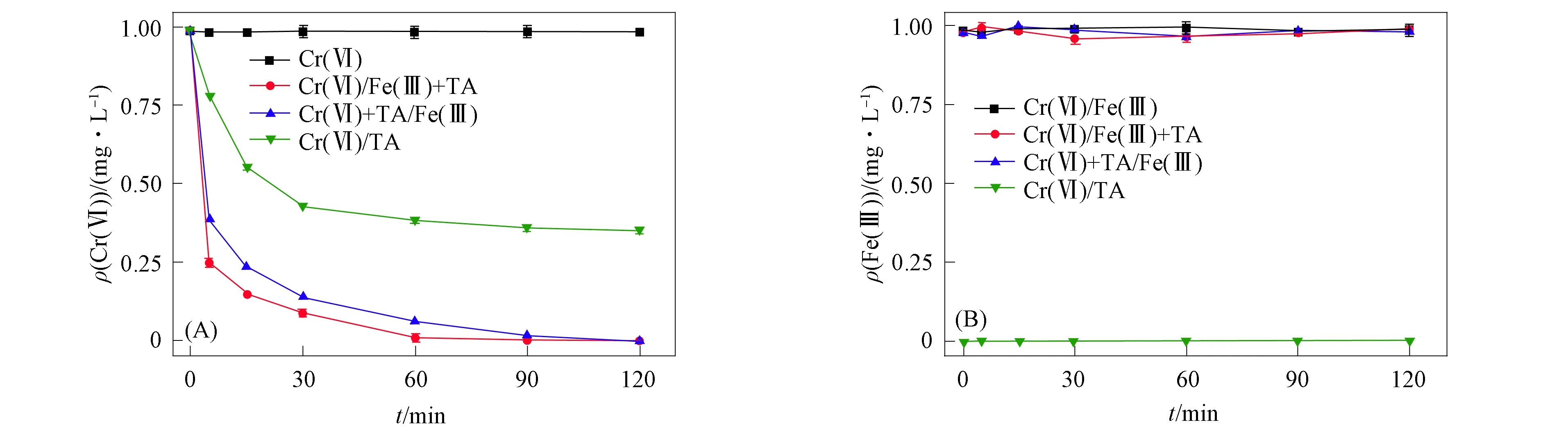
图1 Fe(Ⅲ)对TA原位修复Cr(Ⅵ)污染含水层效果的影响(A)及反应体系中Fe(Ⅲ)质量浓度的变化(B)Fig.1 Effect of Fe(Ⅲ) on in situ remediation of Cr(Ⅵ) contaminated aquifer by TA (A) and change of Fe(Ⅲ) mass concentration in reaction system (B)
将TA与Fe(Ⅲ)混合后加入Cr(Ⅵ)污染地下水中同样可使Cr(Ⅵ)的还原率达到99.9%. 其中, TA与Fe(Ⅲ)反应先于TA与Cr(Ⅵ)反应, TA先与Fe(Ⅲ)形成稳定的配合物, 之后再通过电子转移的方式将Cr(Ⅵ)还原. TA与Cr(Ⅵ)的配合分为4个步骤[20]: 1) 铬酸盐与TA酯化; 2) TA将Cr(Ⅵ)还原为Cr(Ⅲ); 3) 鞣质分子氧化形成醌; 4) 静电吸引Cr3+吸附. 因此, TA与Cr(Ⅵ)的配合远大于TA与Fe(Ⅲ)的配合难度, 从而保证了体系中的Fe(Ⅲ)能起到电子穿梭体的作用, 对TA还原Cr(Ⅵ)进行强化.
2.2 TA和Fe(Ⅲ)初始质量浓度对修复Cr(Ⅵ)污染含水层效能的影响
当反应体系中TA的初始质量浓度发生变化时, Cr(Ⅵ)的还原速率、 还原率以及Fe(Ⅲ)的质量浓度均发生变化, 如图2所示.
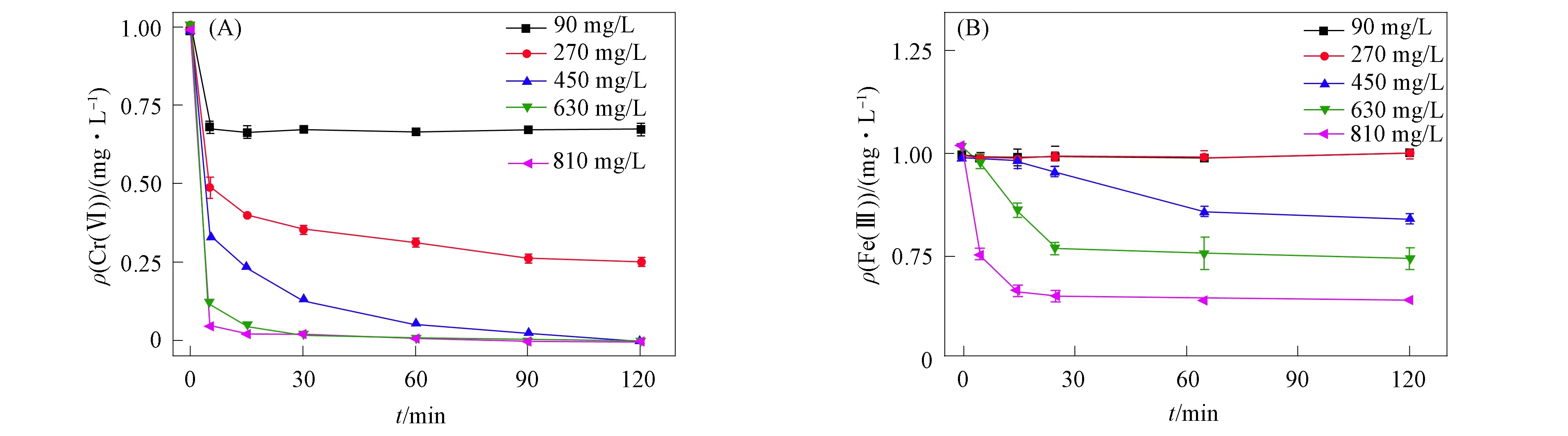
图2 初始质量浓度对Fe(Ⅲ)强化TA原位修复Cr(Ⅵ)污染含水层效果的影响(A)及 反应体系中Fe(Ⅲ)质量浓度的变化(B)Fig.2 Effect of TA initial mass concentration on in situ remediation of Cr(Ⅵ) contaminated aquifer by Fe(Ⅲ) enhanced TA (A) and change of Fe(Ⅲ) mass concentration in reaction system (B)
由图2可见, 当保持Fe(Ⅲ)初始质量浓度不变而增大TA的初始质量浓度时, Cr(Ⅵ)的还原速率增大, Cr(Ⅵ)的还原率也上升. 当TA的初始质量浓度为450 mg/L时, Cr(Ⅵ)的还原率可达99.9%, 此时m(TA)∶m(Cr(Ⅵ))=9∶1, 即n(TA)∶n(Cr(Ⅵ))=1∶3. 可见, TA对Cr(Ⅵ)的还原能力取决于TA与Fe(Ⅲ)配位的酚羟基数量. 根据化学计量比计算可知, 1 mol的TA中含酚羟基数量约为25 mol, 但参与Cr(Ⅵ)还原反应的酚羟基数量仅约为10 mol. 在1 mol TA中, 共有5 mol的邻苯三酚结构, 其中每个邻苯三酚结构中可与Fe(Ⅲ)进行配位酚羟基仅有两个处于邻位的酚羟基[20]. 当酚羟基和Fe(Ⅲ)形成配位键后, 电子更容易由TA转移至Cr(Ⅵ)上. 未与Fe(Ⅲ)形成配位的酚羟基则很难与Cr(Ⅵ)发生反应.
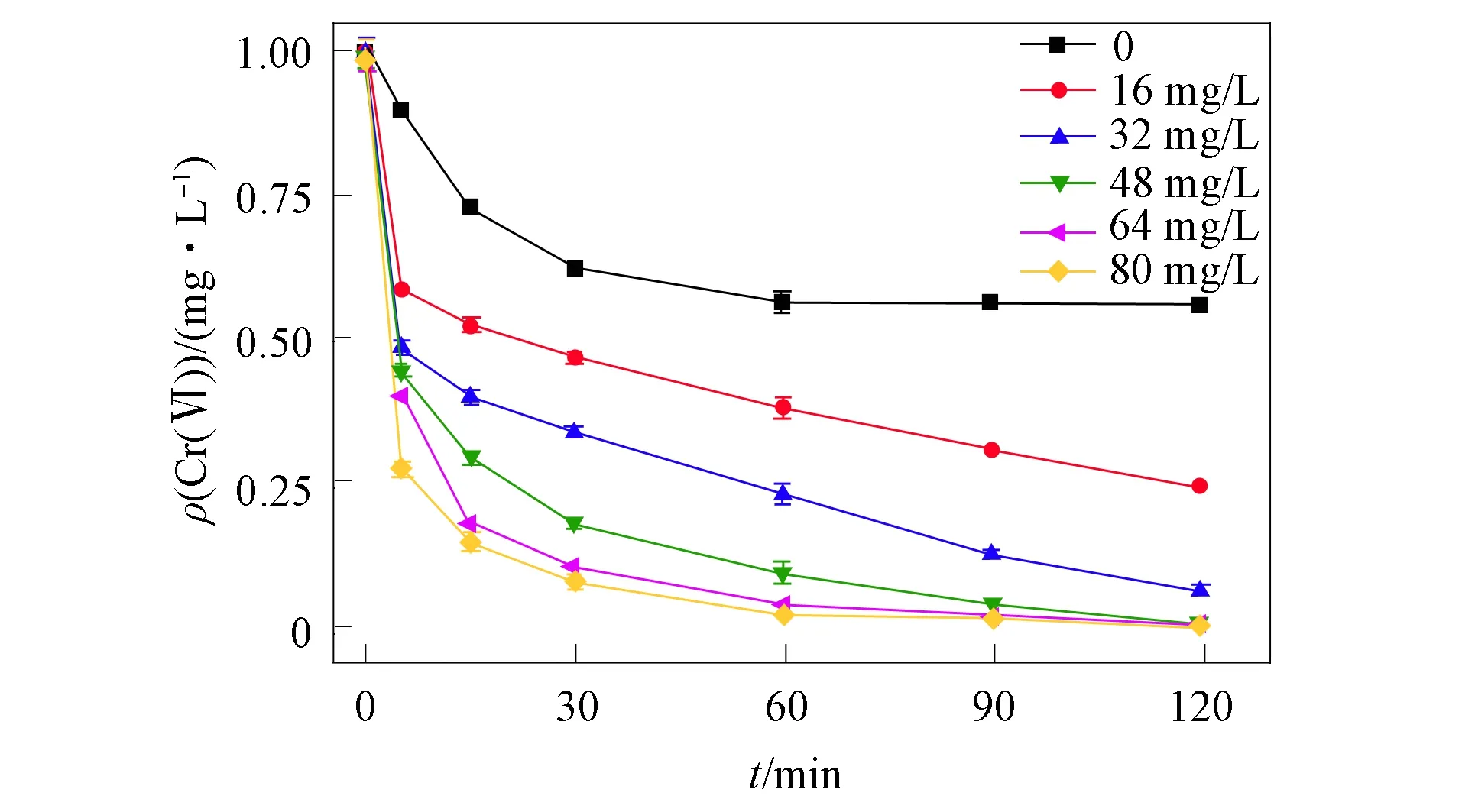
图3 Fe(Ⅲ)初始质量浓度对Fe(Ⅲ)强化TA原位 修复Cr(Ⅵ)污染含水层效果的影响Fig.3 Effect of Fe(Ⅲ) initial mass concentration on in situ remediation of Cr(Ⅵ) contaminated aquifer by Fe(Ⅲ) enhanced TA
当反应体系中Fe(Ⅲ)的初始质量浓度发生变化时, Cr(Ⅵ)的还原速率发生变化, 如图3所示. 由图3可见, TA的初始质量浓度保持不变, 若增加Fe(Ⅲ)的初始质量浓度, 则Cr(Ⅵ)的还原速率明显提高. 但当m(Fe(Ⅲ))∶m(TA)>1∶14, 即n(Fe(Ⅲ))∶n(TA)>5∶3时, 再增加Fe(Ⅲ)的初始质量浓度对Cr(Ⅵ)还原速率的提升并不明显. 结果表明, Fe(Ⅲ)对TA还原Cr(Ⅵ)的速率受Fe(Ⅲ)/TA配合物稳定性的影响. 根据化学计量比计算, 在该配比下, TA分子与Fe(Ⅲ)离子的数量比约为3∶5. 文献[24]研究表明, 当角频率为35 rad/s时, 随着Fe3+数量的增加, TA/Fe(Ⅲ)配合物的存储容量先增加后略降低. 当n(TA)∶n(Fe(Ⅲ))=3∶5时, 储存容量达到最大值, 此时TA/Fe3+配合物最稳定, 因此Fe(Ⅲ)可起到较好的强化作用.
2.3 铁形态与铬形态分析
图4为Fe(Ⅲ)强化TA原位修复Cr(Ⅵ)污染含水层过程中Cr(Ⅵ)的形态变化; 图5为Fe(Ⅲ)强化TA原位修复Cr(Ⅵ)污染含水层过程中Fe(Ⅲ)的形态变化; 图6为TA与Fe(Ⅲ)单独反应过程中Fe(Ⅲ)的形态变化.
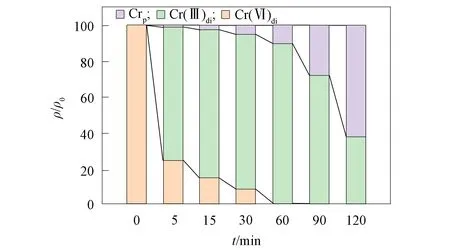
图4 Fe(Ⅲ)强化TA原位修复Cr(Ⅵ)污染 含水层过程中Cr(Ⅵ)的形态变化Fig.4 Morphological changes of Cr(Ⅵ) in process of in situ remediation of Cr(Ⅵ) contaminated aquifer by Fe(Ⅲ) enhanced TA

图5 Fe(Ⅲ)强化TA原位修复Cr(Ⅵ)污染 含水层过程中Fe(Ⅲ)的形态变化Fig.5 Morphological changes of Fe(Ⅲ) in process of in situ remediation of Cr(Ⅵ) contaminated aquifer by Fe(Ⅲ) enhanced TA
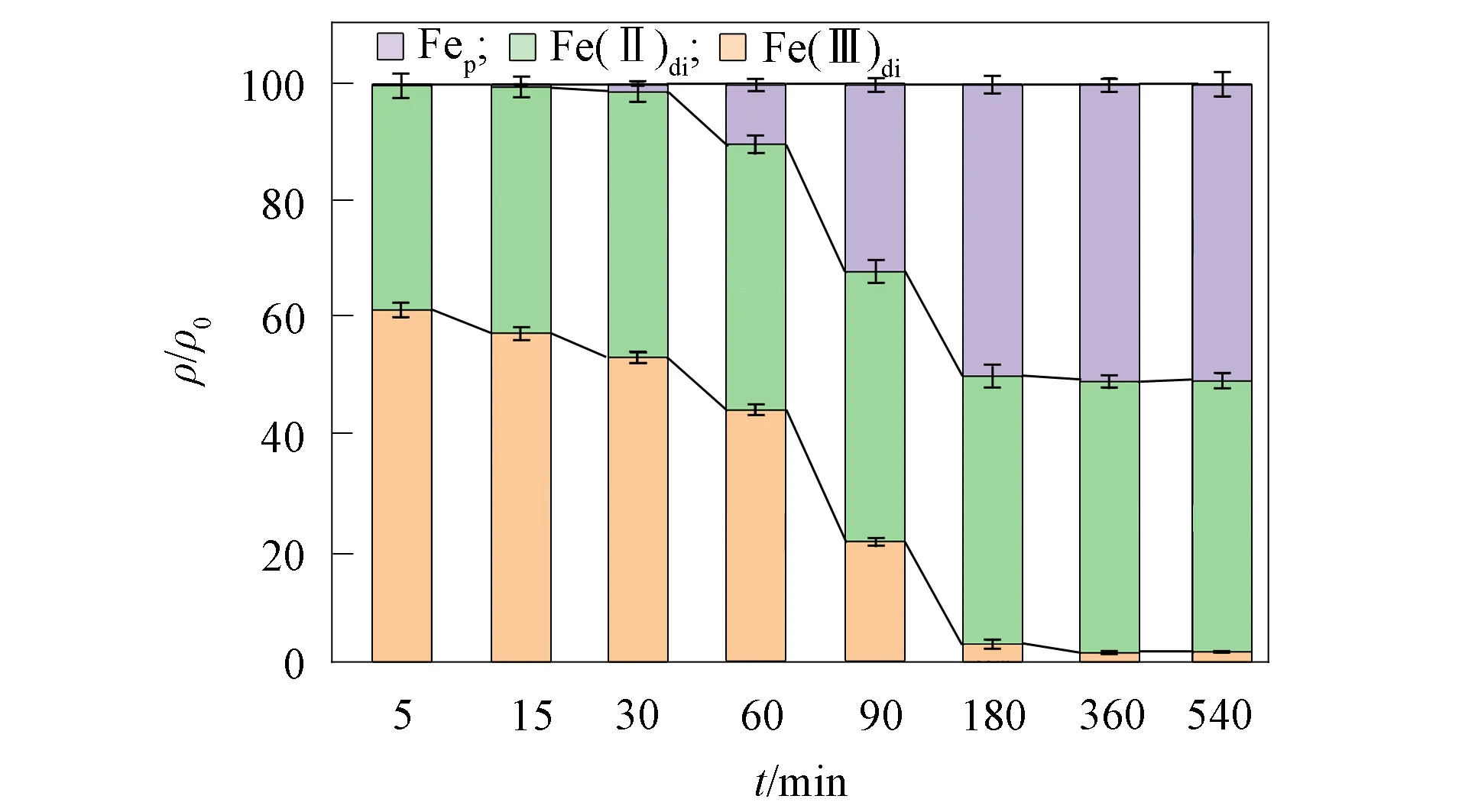
图6 TA与Fe(Ⅲ)单独反应过程中 Fe(Ⅲ)的形态变化Fig.6 Morphological changes of Fe(Ⅲ) in process of reaction between TA and Fe(Ⅲ)
由图4可见, Fe(Ⅲ)/TA体系还原Cr(Ⅵ)反应开始后5 min内, 体系中Cr(Ⅵ)的质量浓度迅速降低. 随着反应进行, 少量Cr从溶液中析出形成沉淀. 当Cr(Ⅵ)被完全还原后, Crp的析出速率明显增加. 在120 min时, 溶液中仅存在Cr(Ⅲ), 其余Cr均以颗粒相存在. 由图5可见, 该过程中, 前60 min内Fe主要以Fe(Ⅲ)的形式存在, 在反应60 min后, Cr(Ⅵ)已被完全还原为Cr(Ⅲ), Fe(Ⅱ)di和Fep开始生成. 随着反应时间的增加, Fe(Ⅲ)的质量浓度显著降低, Fe(Ⅱ)di和Fep的生成量不断升高. 相比于单独的TA/Fe(Ⅲ)反应体系(图6), 当体系中存在Cr(Ⅵ)时, Fep出现的时间相对较晚, 但最终产量更高.
上述结果表明, Fe(Ⅲ)可提高TA还原Cr(Ⅵ)的速率以及最终效率, 并可有效去除体系中生成的Cr(Ⅲ). 文献[25]研究表明, 少量的Fe(Ⅱ)可有效促进Fe(OH)3凝胶转化为FeOOH. 在酸性厌氧还原条件下, 酸根离子与Fe(Ⅱ)共存可促进γ-FeOOH的溶解和再结晶. 当加入其他金属离子(如Ti(Ⅳ),Cr(Ⅲ),Cu(Ⅱ))时,γ-FeOOH可转变为α-FeOOH, 晶型更稳定[26-28]. 有机酸及其盐类(如柠檬酸盐和草酸盐)可通过配位或螯合作用与Fe(Ⅲ)共沉淀, 而大分子有机聚合物与Fe(Ⅲ)的共沉淀作用更明显. 孙振亚等[29]研究表明, 生物矿化针铁矿对Cr(Ⅲ)和Cr(Ⅵ)均有较好的吸附作用, 这也解释了Cr(Ⅲ)总量明显降低的原因.
对经过干燥的颗粒样品进行X射线光电子能谱(XPS)分析, 结果如图7所示. 由图7(A)可见, 在反应120 min后, 在577.45,587.30 eV处出现两个谱峰, 分别归属于Cr2O3的Cr 2p3/2轨道和Cr 2p1/2轨道, 由此可判定颗粒物中的Cr主要以Cr(OH)3的形态存在. 表明在TA/Fe(Ⅲ)体系加入 Cr(Ⅵ)污染地下水中后, Cr(Ⅵ)被完全还原为Cr(Ⅲ), 之后Cr(Ⅲ)以沉淀的方式从溶液中析出, 使体系中总铬含量明显降低. 由图7(B)可见, Fe 2p3/2区反褶积显示出两个峰值, 711.3 eV处的峰归属于Fe 2p3/2, 724.9 eV处的峰归属于Fe 2p1/2. 719.3,732.9 eV处的峰值是两个卫星峰值. 与FeOOH材料的XPS表征谱峰相同[29-31].
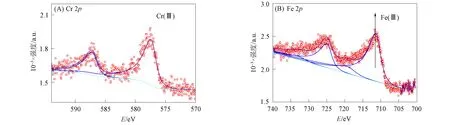
图7 颗粒相物质表面Cr(A)和Fe(B)的化学状态Fig.7 Chemical states of Cr (A) and Fe (B) on surface of particulate matters
2.4 官能团分析
用紫外-可见光分光光度计对Fe(Ⅲ),TA,Cr(Ⅵ),TA/Fe(Ⅲ)体系以及TA/Fe(Ⅲ)+Cr(Ⅵ)溶液进行扫描, 结果如图8所示. 由图8(A)可见: TA在271 nm处的吸收峰在加入Fe(Ⅲ)后消失, 表明TA上的酚羟基被消耗; TA/Fe(Ⅲ)在230 nm处新的吸收峰归属于酚羟基被氧化后生成的醌类以及半醌类物质. 由图8(B)可见, 当Cr(Ⅵ)加入TA/Fe(Ⅲ)体系时, 属于醌类物质的吸收峰强度增加[32-33], 表明半醌类物质被彻底氧化为醌类物质, 同时电子转移给Cr(Ⅵ)使其被还原为Cr(Ⅲ). 反应方程式如下:

(5)

(6)
(7)

(8)
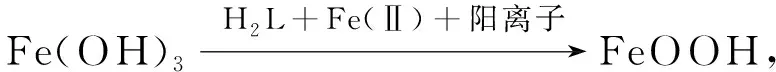
(9)
(10)
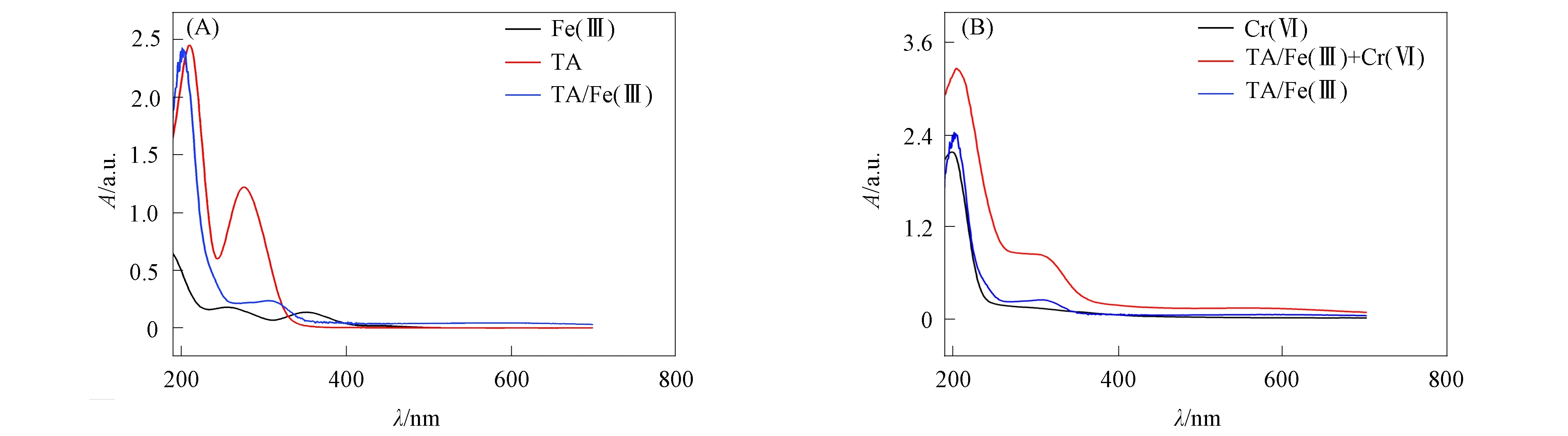
图8 Fe(Ⅲ),TA和TA/Fe(Ⅲ)体系(A)及Cr(Ⅵ),TA/Fe(Ⅲ)体系 和TA/Fe(Ⅲ)+Cr(Ⅵ)体系(B)的紫外-可见光谱 Fig.8 UV-Vis spectra of Fe(Ⅲ),TA and TA/Fe(Ⅲ) system (A) and Cr(Ⅵ),TA/Fe(Ⅲ) system and TA/Fe(Ⅲ)+Cr(Ⅵ) system (B)
2.5 TA/Fe(Ⅲ)复合体系修复Cr(Ⅵ)污染地下水机理分析
上述结果表明, Fe(Ⅲ)被还原生成的Fe(Ⅱ)在TA/Fe(Ⅲ)体系中还原Cr(Ⅵ)和去除Crtot中具有重要作用. Fe(Ⅱ)作为一种传统的还原剂, 对Cr(Ⅵ)的还原效果较好, 而加入TA可使Fe(Ⅱ)被消耗后再生. 同时, Fe(Ⅲ)在反应过程中不仅作为Fe(Ⅱ)的来源, 还能与TA形成稳定的配合物. Fe(Ⅲ)与TA的氧配位体形成强配位体, 作为电子穿梭体, 极大提高了酚羟基向Cr(Ⅵ)的电子转移效率, 改变了TA还原Cr(Ⅵ)的基本反应途径.
图9为TA/Fe(Ⅲ)体系还原反应途径. Cr(Ⅵ)的去除过程可分为氧化还原和共沉淀两部分. 首先, Fe(Ⅲ)作为电子穿梭体提高了电子转移效率, 从而增强了TA还原Cr(Ⅵ)的能力. 当TA过量时, Fe的氧化还原过程不断将电子从TA转移至Cr(Ⅵ), 该过程中Cr(Ⅵ)被还原为Cr(Ⅲ), 酚羟基被氧化为醌. 其次, Fe(Ⅲ)水解生成Fe(OH)3胶体, 在Cr(Ⅲ)、 Fe(Ⅱ)和有机酸存在下Fe(OH)3溶解再结晶成FeOOH. 最后, FeOOH通过对Cr(Ⅲ)的吸附能力将Cr(Ⅲ)固定, 并形成稳定的氧化物沉淀, 从环境中去除总铬. 在反应过程中, 由于反应体系的pH值未变, 约为4.0, 因此无需引入碱性物质调节环境pH值, 促使Cr(Ⅲ)沉淀.
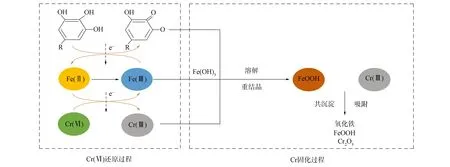
图9 TA/Fe(Ⅲ)体系同步还原Cr(Ⅵ)并去除Crtot的机理Fig.9 Mechanism of simultaneous reduction of Cr(Ⅵ) and removal of Crtot by TA/Fe(Ⅲ) system
3 结 论
本文通过批处理实验研究Fe(Ⅲ)强化TA原位修复含水层Cr(Ⅵ)污染的反应机理, 分析了TA初始质量浓度与Fe(Ⅲ)初始质量浓度对Fe(Ⅲ)强化TA原位修复含水层Cr(Ⅵ)污染效果的影响, 研究了Fe(Ⅲ)强化TA原位修复含水层Cr(Ⅵ)污染过程中Fe和Cr的存在形态, 并给出了Fe(Ⅲ)强化TA原位修复含水层Cr(Ⅵ)污染的反应方程式. 可得如下结论:
1) TA中的酚羟基作为电子供体, Cr(Ⅵ)作为电子受体, Fe(Ⅲ)作为电子穿梭体提高了电子转移的效率;
2) 还原1 mol的Cr(Ⅵ)最少需要9 mol的TA;
3) 当n(TA)∶n(Fe(Ⅲ))=3∶5时, Fe(Ⅲ)对TA原位修复含水层Cr(Ⅵ)污染效能的强化效果最好;
4) Fe(Ⅲ)在Cr(Ⅵ)被还原后与TA发生反应, 并形成稳定的α-FeOOH(针铁矿), 对Cr(Ⅲ)进行吸附, 达到去除总Cr的目的.
