细颗粒污染物PM2.5与慢性肾脏疾病
2021-09-18欧阳彦综述谢静远审校
李 想 欧阳彦 综述 谢静远 审校
空气污染作为公共问题一直备受重视,全球每年超过300万例的死亡以及7 600万例的健康寿命损失与其密切相关[1-2]。空气污染物包括氮氧化物、硫氧化物、一氧化碳、细颗粒物(particulate matter,PM)等。颗粒物直径≤10 μm的PM可沉积于肺泡,从而危害人体健康,被称为可吸入PM(PM10)。PM10根据根据直径进一步细分为粗颗粒(PM10-2.5,2.5~10 μm)、细颗粒物(PM2.5,≤2.5 μm)和超细颗粒物(PM0.1,≤0.1 μm)。可通过地面监测站或卫星遥感监测空气中PM的浓度。
国内外研究显示,PM的暴露导致每年约300万人口过早死亡,占全球死亡人数的5.86%[3]。其中,PM2.5可穿过肺泡气血屏障进入肺循环,到达包括肾脏在内脏器并造成损伤。PM2.5被认为是导致疾病发生率和死亡率上升的最主要的空气污染物之一[4]。 Liu等[5]从24个国家或地区的652个城市收集了日死亡率和日空气污染数据,浓度-响应曲线显示PM2.5浓度每增加10 μ g/m3,全因死亡率增加0.68%(95%CI 0.59~0.77)。
慢性肾脏病(CKD)在我国成年人中患病率约为10.8%[6-7]。全球CKD负担研究显示,2017年全球CKD患者约6.97亿人,同年因CKD致死的人数超过120万[8]。大量研究表明,多种环境污染介导了肾脏损伤,其中空气污染特别是PM2.5的暴露与CKD密切相关[9]。Bowe等[10]构建PM2.5暴露-反应模型并估算CKD的全球经济负担,发现PM2.5引起的CKD全球负担极重:暴露反应函数模型显示,2017年全球PM2.5暴露导致3 284 358.2例(95%CI 2 800 710.5~3 747 046.1例)CKD事件;导致6 593 134.6例(95%CI 5 705 180.4~7 436 870.1例)伤残调整寿命年(DALYs)下降事件,并导致211 019.2例(95%CI 184 292.5~236 520.4例)死亡事件。为进一步探讨PM2.5与CKD的关联,本文就这一领域的新进展作一综述。
PM2.5与肾脏损伤相关
印度北部一项关于PM2.5暴露的研究[11]纳入94名厨房工作者,并以94名办公室工作人员作为对照,发现厨房PM2.5浓度远高于室内PM2.5浓度(81.3±51.2 μg/m3vs34.7±12.6 μg/m3,P<0.001),厨房工作人员的微量蛋白尿患病率显著高于对照组(85.11%vs22.3%,P<0.001)。该研究提示厨房PM2.5可能与微量蛋白尿的发生有关。Chuang等[12]对66名焊接工人和12名办公室工人进行研究,使用实时粉尘检测仪测量个体暴露PM2.5浓度。该研究发现,焊接工人的 PM2.5平均暴露水平显著高于办公室工人(50 μg/m3vs27 μg/m3,P<0.001),且焊接烟雾暴露后工人尿液肾损伤分子1(KIM-1)和中性粒细胞明胶酶相关脂质运载蛋白(NGAL)水平增加,提示 PM2.5暴露导致肾损的机制可能与肾小管受损有关。
PM2.5与CKD发生Blum等[13]纳入1987年~1990年来自美国四个地区的10 997名社区居民,发现年平均PM2.5浓度与尿蛋白水平和CKD发病风险存在正相关。经过人口学参数、社会经济状况和临床协变量的多因素校正后,年平均PM2.5浓度每升高1 μg/m3, CKD风险增加5%(HR=1.05,95%CI 1.01~1.10),同时尿白蛋白肌酐比值升高6%(95%CI 2.6%~10.7%)。Chan等[14]开展一项队列研究以分析PM2.5长期暴露与CKD发病的关联,2001年~2014年间纳入年龄≥20岁且随访>3年的非CKD中国台湾居民100 629名。在随访的过程中4 046例发生CKD。使用卫星时空模型估计患者居住地PM2.5浓度,并应用年龄、性别、教育程度、吸烟、饮酒、体质量指数等参数校正后,发现PM2.5浓度每升高10 μg/m3,CKD的患病风险增加6%(HR=1.06, 95%CI 1.02~1.10)。
Xu等[15]纳入中国282个城市的938家医院,收集2004年~2014年间71 151例的肾活检病理报告。在校正年龄,性别等影响因素后发现,膜性肾病的发生率以每年13%在增长。全国不同区域PM2.5浓度与膜性肾病发病率的关联研究发现,PM2.5>70 μg/m3的地区,PM2.5浓度每上升10 μg/m3,膜性肾病的发病风险上升14%(HR=1.14, 95%CI 1.10~1.18)。这项研究提示高水平的PM2.5暴露导致膜性肾病的发病风险增加。
PM2.5与CKD进展为明确PM2.5与CKD预后的相关性,Bowe等[16]开展一项纳入2 482 737名美国退伍军人的观察队列,中位随访时间为8.52年。调整协变量(包括年龄、种族、性别、体质量指数及吸烟状况等)后,PM2.5浓度每升高10 μg/m3,CKD风险增加27%(HR=1.27,95%CI 1.17~1.38),终末期肾病(ESRD)风险增加26%(HR=1.26,95%CI 1.17~1.35),估算的肾小球滤过率(eGFR)减退风险增加28%(HR=1.28,95%CI 1.18~1.39)。另一项来自中国台湾的队列研究[17]纳入6 628例CKD患者,研究者发现PM2.5水平每增加7.8 μg/m3,进展至ESRD风险增加19%(HR=1.19,95%CI 1.08~1.31)。Chen等[18]为分析空气污染与老年人CKD的关联,纳入8 497名年龄>65岁的台北居民。研究发现PM2.5吸光度每增加0.4×10-5/m导致 CKD患病风险升高12.6%,(OR=1.126,95%CI 1.057~1.199),CKD进展风险增高11.4%(OR=1.114,95%CI 1.051~1.181)。上述研究在健康人群以及肾脏疾病人群中均证实PM2.5暴露与肾衰竭的发生有关。
PM2.5所致肾脏损伤病理改变
为研究PM2.5对全身器官的影响,严超等[19]将40只SD大鼠随机分为三组,生理盐水对照组10只,低剂量[2 μg/100(g·d)]和高剂量[16 μg/100(g·d)]PM2.5长期暴露组各15只,连续暴露60d。结果显示高剂量组大鼠体重明显减轻,大鼠肺、心、肝、脾、肾等脏器均有明显的炎症反应,高剂量组肾组织中,部分肾小球毛细血管腔开放不佳、肾小球动脉硬化改变、部分包囊轻微扩张以及少量包囊破裂。Yan等[20]探究PM2.5对糖尿病大鼠靶器官的影响,用链脲霉素构建糖尿病大鼠模型,并将大鼠暴露于平均PM2.5浓度为13.30 μg/m3的环境中达16周。免疫组化结果显示,糖尿病大鼠的肾小球硬化增加且肾小管损伤加重。周晨曦等[21]通过建立大鼠PM2.5经呼吸道亚慢性染毒模型,病理显示大鼠肾脏肾小球轻度萎缩,伴炎细胞浸润,血管扩张充血,局部近曲小管和远曲小管中度肿胀。这些研究均基于动物模型,尚缺乏人体中PM2.5暴露与肾脏病理损伤的研究。
PM2.5参与肾脏损伤机制
PM2.5可通过多种途径导致肾脏损伤(图1)。
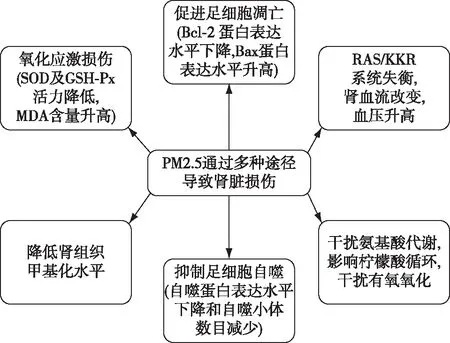
图1 PM2.5对肾脏的损伤机制[21-29]
氧化应激损伤周晨曦等[21]通过建立大鼠PM2.5经呼吸道亚慢性染毒模型,探究PM2.5肾脏损伤机制,研究者将大鼠肾脏与生理盐水固定比例混合制备组织匀浆,离心后取上清液,应用相应试剂盒检测大鼠肾脏超氧化物歧化酶(SOD)、谷胱甘肽过氧化物酶(GSH-Px)以及丙二醛(MDA)含量,与对照组比较,PM2.5染毒组大鼠肾脏组织的SOD及GSH-Px含量均降低,而MDA含量升高;采用免疫组化的方法检测Bcl-2及Bax蛋白的表达情况,发现Bcl-2蛋白主要表达于肾小管,Bax蛋白在肾小球及肾小管均有表达,与对照组相比,Bcl-2蛋白表达水平下降,Bax蛋白表达水平升高;应用原位缺口末端标记法(TUNEL)检测肾脏细胞凋亡情况,发现PM2.5染毒组大鼠肾脏细胞凋亡率高于对照组(25.93%±3.68%vs35.15%±4.39%,P<0.05)。结果显示,PM2.5可致大鼠肾脏发生明显的氧化损伤效应,从而导致细胞凋亡。采油机排气颗粒(DEP)是城市地区PM2.5和直径≤0.1 μm的超细颗粒的主要来源之一。Nemmar等[22]评估长期暴露于DEP对腺嘌呤诱发的慢性肾衰竭的作用。其研究发现,长期暴露于DEP的慢性肾衰竭大鼠体内,肿瘤坏死因子α、脂质过氧化和活性氧显著升高;彗星实验显示DEP暴露的慢性肾衰竭大鼠肾脏细胞DNA迁移率较对照组增加(P<0.05),提示DNA损伤增加;大鼠肾脏组织匀浆检测显示过氧化氢酶等抗氧化酶含量降低。组织病理学检查显示DEP暴露的慢性肾衰竭大鼠肾脏坏死细胞计数增加,肾小球硬化以及肾小管纤维化程度较对照组加重。研究显示长期暴露于DEP会加重腺嘌呤诱发的慢性肾衰竭的大鼠的氧化应激、炎症和DNA损伤。
肾脏血流改变Suleimani等[23]研究分析DEP对慢性肾脏病大鼠的血管影响,发现DEF暴露大鼠的肾脏血流量(RBF)显著下降:与对照组相比,DEF暴露组大鼠的收缩压及舒张压升高、去氧肾上腺素诱导的血管收缩均更为明显。该研究进一步分析阿拉伯树胶(GA)对血管损伤的改善作用,发现对于DEP暴露导致的血管损伤,GA并无改善作用。Aztatzi-Aguilar等[24]既往研究发现PM2.5暴露的大鼠的心、肺组织中,肾素血管紧张素系统(RAS)和缓激肽(KKR)系统标志物表达增加。研究者进一步分析PM2.5导致早期肾脏受累,结果显示[25]与对照组相比,PM2.5暴露组大鼠的血肌酐升高(0.6 mg/dlvs0.7 mg/dl,P=0.03),肾组织中RAS相关基因(AT1R 和Ace)和KKR相关基因(B1R和KLK-1)编码蛋白均表达上调,免疫组化显示其肾间质纤维化更重。提出亚慢性的PM2.5暴露导致早期肾脏损害,可能与RAS/KKR失衡有关。
足细胞损伤肾脏足细胞即肾小球脏层上皮细胞,是一种终末分化细胞,数量少且分化能力弱,可作为肾脏疾病进展或肾脏衰老的指标之一[30]。多环芳烃(PAH)是PM2.5中常见的有机化学物质,而苯并[b]荧蒽(BbF)是PAH最主要的成分。Zhang等[26]为研究BbF对肾脏足细胞的损伤机制,应用MPC小鼠肾脏足细胞做体外实验,结果发现随着BbF浓度上升,小鼠MPC细胞存活率下降,当BbF浓度升至25 μg/ml,暴露组MPC细胞存活率降至52.0%±4.1%。进一步分析发现,随着暴露时间延长,MPC细胞存活率下降——BbF作用72h时,MPC细胞存活率下降至52.4%。这些结果提示BbF以剂量和时间依赖的方式诱导足细胞损伤,通过透射电镜观察发现暴露组自噬蛋白表达水平下降和自噬小体数目减少,且呈时间和剂量依赖性,提示BbF可能通过抑制足细胞的自噬导致肾脏损伤。
代谢损伤为研究PM2.5的毒性机制,Zhang等[27]运用基于H核磁共振(NMR)代谢组学的方法,探讨气管内滴注PM2.5后大鼠的内源性代谢变化以及可能影响的代谢途径,并使用H-NMR技术结合多元统计分析系统地分析了大鼠血清和尿液的代谢物变化,发现PM2.5暴露大鼠血清中乳酸、丙氨酸、二甲基甘氨酸、肌酸、甘氨酸和组氨酸的水平显著降低,尿液中柠檬酸、精氨酸、马尿酸盐、尿囊素的水平升高,而苏氨酸、乳酸、丙氨酸、乙酸盐、琥珀酸、三甲胺和甲酸盐的水平降低。观察到PM2.5主要影响的代谢途径是甘氨酸、丝氨酸和苏氨酸代谢,柠檬酸循环和氨代谢等。代谢组学分析表明,暴露于PM2.5会干扰氨基酸代谢,并影响柠檬酸循环从而影响糖有氧氧化。
DNA甲基化DNA甲基化是最早发现的表观遗传修饰途径之一,其中5-甲基胞嘧啶(5-mc)是最主要的表观遗传标志物之一,也是研究最为广泛的表观遗传修饰形式。Li等[28]为研究PM2.5暴露对不同组织DNA甲基化的影响,用小鼠构建PM2.5暴露模型,将实验小鼠分为急性暴露组和慢性暴露组。急性组小鼠饲养于PM2.5浓度为271.8±86.8 mg/m3的高PM2.5浓度环境中暴露24h,对照组饲养于经过PM2.5过滤的环境下PM2.5浓度为19.8±9.0 μg/m3;慢性暴露组:小鼠饲养于PM2.5浓度为91.3±84.8 μg/m3的环境中,持续暴露140d,对照组饲养于经PM2.5过滤的环境中,PM2.5浓度为17.9±7.8 μg/m3,结果发现无论是急性或者慢性PM2.5暴露,小鼠肾组织中5-mc水平与对照组相比,虽然差异无统计学意义,但仍可见下降趋势,提示DNA甲基化水平的下降可能参与了PM2.5导致的肾脏损伤。
为探究PM2.5暴露对器官或组织产生的遗传毒性,Antonio等[29]将实验小鼠置于PM2.5浓度为682±532 μg/m3环境下暴露1 h/d,暴露5 d/周,持续3个月构建慢性PM2.5暴露模型,对照组呼吸环境空气,结果发现实验组小鼠肾组织中5-mc为4.20%±0.37%,对照组为4.84%±0.24%,虽然与对照组相比差异无统计学意义,但仍能看到下降趋势;该研究还发现PM2.5暴露组小鼠肾组织8-oxdGuo(是一种主要的DNA氧化损伤产物)水平上升,提示DNA甲基化水平的下降与DNA氧化损伤可能参与了PM2.5暴露导致的肾脏损伤。
展 望
PM2.5的过量暴露,会增加肾脏疾病的发生率和死亡率。PM2.5导致肾脏受损的机制十分复杂,主要通过氧化应激、足细胞损伤、肾血流改变、代谢以及DNA甲基化修饰等方面导致肾脏受损。但目前PM2.5暴露参与CKD发生发展中的具体机制暂未完全明确,特别是与PM2.5导致肾脏损伤的信号通路、细胞因子及甲基化修饰等的具体机制仍待进一步探索,此外,还应该对遗传背景与PM2.5肾脏损伤的易感性,以及可用于临床的生物标记物进行深入研究,这些研究结果将对于防治大气污染对人类健康的影响起到关键作用。
