紫外光谱对液相色谱法对5种食品添加剂的定性研究
2021-09-15张房宇
张房宇
(广州市从化质量技术监督检测所 广东广州 510925)
1 前言
液相色谱法具有应用范围广、样品前处理简单、高效高速等优点。十多年来,相关学者对液相色谱法同时检测食品中2种及以上添加剂的研究有很多,其中,紫外光度检测器是液相色谱中应用最广泛的检测器。
同时测定食品中山梨酸、苯甲酸、糖精钠、安赛蜜、脱氢乙酸的高效液相色谱法[1-15]发展迅速,被广泛采用。但是在实际检测中,由于被测样品中成份复杂,大多只经简单预处理,微孔膜过滤后直接注入液相色谱仪,样品的基质及pH值等因素影响待测物在色谱柱里的吸附洗脱过程,经色谱柱分离后得到的色谱峰往往容易发生漂移,造成根据色谱峰的保留时间进行定性判定的困难。
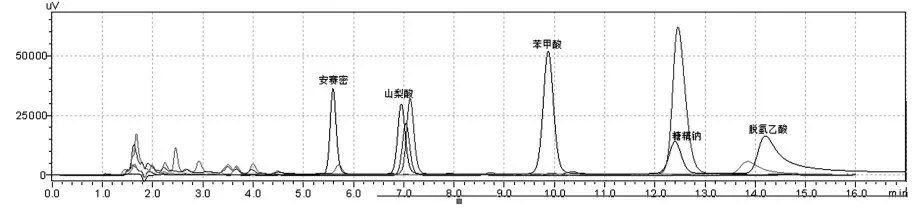
图1 一个混合标准样品与三个待测样品在230 nm波长下的液相色谱重叠图
而国外通常采用SPE固相萃取柱对样品进行前处理,采用固相填料对样品组分的选择性吸附、选择性洗脱的方式,实现对待测物的富集、分离、纯化,降低样品基质干扰,提高检测灵敏度和准确度。但是由于SPE固相萃取柱的采购成本较高,我国并未普遍使用此方法。
目前该液相色谱国标检测方法[18-20]仍旧是依据保留时间进行定性的,除了改用气相色谱法、改变流动相[18]或是液质联用[2,13]的其它定性分析验证方法外,紫外光谱定性分析在实际检测应用过程中的深入研究未见报道。本文不作检测方法的讨论,只在选定的色谱条件下,利用紫外二极管阵列检测器对经过色谱柱分离出来的标准物质山梨酸、苯甲酸、糖精钠、安赛蜜、脱氢乙酸的色谱峰进行光谱扫描,依据分子吸收光谱原理对其相应的紫外光谱图进行解读,进一步探讨相应的标准物质和待测物的紫外光谱图在液相色谱法中定性的应用,以期对依据保留时间进行定性的方法作为相互验证的补充。
2 实验
2.1 试剂
安赛蜜(纯度≥99.0%);苯甲酸(纯度≥99.5%);山梨酸(纯度≥99%);糖精钠(纯度≥99.0%);脱氢乙酸(纯度≥98.0%);乙酸铵(天津光复,分析纯);无水甲醇(色谱纯;超纯水,自制);0.45μm微孔过滤膜(津腾);亚铁氰化钾(国药集团,分析纯);乙酸锌(天津光复,分析纯);碳酸氢钠(天津福晨,优级纯)。
2.2 主要仪器
主要仪器有高效液相色谱仪组合型(SHIMAD ZN):含二元LC-10AT VP泵、CTO-10AS VP柱温箱(带7725(i)型手动进样器)、SPD-M20A二极管陈列检测器、CBM-20A系统控制器;AY-120万分之一电子分析天平(SHIMADZN)。
2.3 溶液的制备
样品处理:精确称取2 g样品置于50 mL离心管中,各加入2 mL亚铁氰化钾(92 g/L)和乙酸锌(92 g/L),混匀,离心5 min后用稀氨水(1+99)调pH值近中性,定容于50 mL容量瓶,经0.45μm微孔滤膜过滤后,待测。
试剂处理:将配制好的流动相0.02 mol/L乙酸铵缓冲液经0.45μm水系微孔滤膜过滤后,脱气后使用;甲醇经0.45μm有机微孔滤膜过滤后,脱气后使用。
安赛蜜、山梨酸、苯甲酸、糖精钠、脱氢乙酸标准储备溶液配制(10 mmol/L):分别精确称取安赛蜜(0.2012 g)、山梨酸(0.1121 g)、苯甲酸(0.1221 g)、糖精钠(0.2052 g)、脱氢乙酸(0.1682 g),其中山梨酸、苯甲酸、脱氢乙酸分别加5mL碳酸氢钠(20 g/L)加热熔解,加纯水定容至100mL;按需用纯水稀释成(0.1~1.0)mmol/L不同浓度水平的系列混合标准溶液。
互联网环境下,成人网络学习资源不断丰富,但与权威的正式的教材书籍相比,网络学习资源更需要学习者的辨别和筛选。在信息传播门槛较低的网络时代,成人学习者不再只是单向地被动地接受和理解信息,他们更要保持清醒的头脑有选择性的加工和整理信息。
2.4 色谱分析条件
色谱柱:Shim-pack Vp-ODS C18(4.6 mm×150 mm);流动相:甲醇-0.02 mol/L乙酸铵缓冲液(体积比为8∶92);流速:1 mL/min;柱温:35℃;进样量5 μL;二极管阵列190~800 nm全扫描。
3 相同液相色谱法条件下的标准物质紫外光谱图解析
3.1 安赛蜜标准物质的紫外光谱图解析
安赛蜜在波长210 nm~250 nm之间有强吸收带(见图2),在最大吸收所对应的波长λmax=226 nm处表明其含有2个双键的共轭单位,主要是由位于4-C=O酮键与位于5-C=C-双键形成含有杂原子的共轭体系,形成新的成键轨道与反键轨道,使与π→π*与n→π*的跃迁能级的能差减小,吸收向长波方向位移。

图2 安赛蜜标准物质的紫外可见光谱图
安赛蜜的化学结构式为:

3.2 苯甲酸标准物质的紫外光谱图解析
苯甲酸在波长210 nm~240 nm之间有一强吸收带,在波长250 nm~280 nm之间有一较弱吸收带,分别称为E2吸收带[16]和B吸收带[16],相应最大吸收所对应的波长λmax=223 nm、λmax=268 nm;苯甲酸的E1、E2吸收带是由苯环结构中3个乙烯的环状共轭系统的跃迁所产生——E1吸收带因为背景吸收,在图3中不能完全分得出来,大约在198 nm附近,因此它的E1吸收带不能做定性依据。苯甲酸是苯环上被助色团-COO-取代,由于n→π*共轭,E2吸收带和K吸收带合并,吸收峰向长波移动。同时苯环上有取代基时,显示精细结构的B吸收带也简单化了——这也可能与极性的流动相有关。

图3 苯甲酸标准溶液的紫外可见光谱图
苯甲酸分子结构式为:

3.3 山梨酸标准物质的紫外光谱图解析
山梨酸在最大吸收所对应的波长λmax=253 nm处有一强吸收带(见图4),这一强吸收带表明有3个共轭单位,是由山梨酸2个双键的π→π*共轭和1个羰基键-COO-的n→π*共轭产生,此共轭体系为典型K吸收带[16]。除此之外,通过放大图4观察到山梨酸在λmax=322 nm处有一微弱吸收带,这一微弱吸收带是由羰基基团-COO-同时发生n→π*跃迁产生,也被称为R(基团)吸收带[16]。

图4 山梨酸标准溶液的紫外可见光谱图
山梨酸钾分子结构式为:

3.4 糖精钠标准物质的紫外光谱图解析
糖精钠在最大吸收所对应的波长λmax=206nm处有一强吸收带(见图5),为糖精钠的邻甲酰基、磺酰亚胺基n→π*跃迁和与苯环共轭(π→π*共轭),K吸收带[16]和E2吸收带重合(峰谷变浅),并略向长波移动(苯的E2吸收带在λmax=204 nm)。同苯甲酸,带苯环的糖精钠在λmax=265 nm处也有B吸收带。

图5 糖精钠标准溶液的紫外可见光谱图
糖精钠分子结构式为:

3.5 脱氢乙酸标准物质的紫外光谱图解析
脱氢乙酸在最大吸收所对应的波长λmax=230 nm处有一强吸收带(见图6),为脱氢乙酸吡喃环中双键π→π*跃迁与酮基团n→π*杂原子共轭产生,是K吸收带。在λmax=292 nm处有一较强吸收带,为脱氢乙酸吡喃环中2,4-酮基与3-羰基中含有σ键和π键,发生π→π*跃迁产生,也是K吸收带。

图6 脱氢乙酸标准溶液的紫外可见光谱图
脱氢乙酸分子结构式为:

4 液相光谱图定性分析讨论与例证
观察以上紫外可见光谱图(图2~6),处于同种极性的流动相中5种标准物质主要是在200 nm~380 nm的近紫外区存在强吸收带,通过不同的吸收带可以推断出化合物的官能团(生色团[8]),为了区分色谱峰的纯度,本文完整保存了可见光谱图。一般来在相同的方法条件下,首先通过液相光谱图比较待测物与标准物质所有的最大吸收所对应的波长λmax,如果λmax不同,则可以判定二者不是同一种物质(如图8、图9),不论是这二者的色谱峰保留时间相同或是接近。反之,二者所有的最大吸收所对应的波长λmax相同,则可认为待测物与标准品具有相同的官能团或生色基团,但不能认为它们是同一物质。

图7 蜂蜜话梅样品的液相色谱图(误检出“苯甲酸”和“山梨酸”,检出糖精钠)

图8 蜂蜜话梅样品中误检出“苯甲酸”的紫外可见光谱图
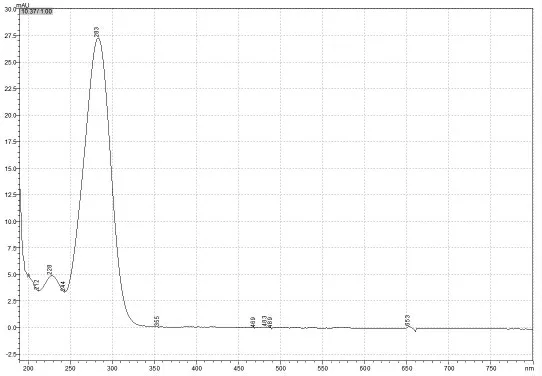
图9 蜂蜜话梅样品中误检出“山梨酸”的紫外可见光谱图
4.1 液相色谱中摩尔吸收系数(εmax)数值比较法
在紫外可见分光光度法中,如果待测物与标准物质的最大吸收所对应的波长λmax相同,还得比较它们的摩尔吸收系数εmax,只有待测物与标准物质的最大吸收所对应的波长λmax和εmax都相同,才可以认为两者是相同物质[16]。根据朗伯-比尔公式A=εbc,c的单位为mol/L,b的单位为cm时;在本文所用的岛津液相色谱仪中SPD-M20A二极管陈列检测器流通池的光程为10 cm,mAU是液相色谱中的吸收度单位,将各自标准物质对应最大吸收所对应的波长λmax处的吸收值代入上式,可以反推算出液相色谱版的摩尔吸收系数εmax=A/bc。通过相同色谱条件下的在(0.1~1.0)mmol/L浓度范围同种标准物质的光谱图数据,计算出的上述5种标准物质分别各自特征波长λmax处的εmax(见表1)。

表1 五种标准物质色谱峰的λmax、εmax与吸收比(A1/A 2)
在液相色谱法中,上述同种标准样品在一定浓度范围内对应λmax处的εmax数值都非常接近,εmax数值随着标准样品浓度的降低缓慢的减小,εmax数值随着标准样品浓度的升高缓慢的增加。只有在2个浓度差别大的同种标准样品的εmax数值才会出现大的偏离,在相同的检测条件下要求标物的浓度要尽可能和未知物的浓度接近。但在检测中由于未知物的浓度未知,通过液相外标法计算出其浓度后再折算成εmax数值与标物εmax数值进行比较的方法,仍有待与其它方法的验证,该方法并不太实用。
从表1可以看出,当εmax数值>1×104数量级时,吸收峰(λmax)处于200 nm~300 nm时,就可以推断此化合物存在共轭双键;苯甲酸和糖精钠B吸收带的εmax数值较弱,山梨酸R吸收带的εmax数值最弱。
4.2 双波长吸收比法
对比任何在紫外-可见光区有吸收的纯物质在2个选定波长下的吸收比为一常数,且与浓度无关[17]。对此,选取具有特征双峰的苯甲酸、山梨酸、糖精钠、脱氢乙酸进行了双波长(λmax)处吸收比(A1/A2)的验证,在相同检测条件下,经计算在(0.1~1.0)mmol/L浓度范围内上述4种标准物质的吸收比(A1/A2)结果见表1。
苯甲酸与糖精钠同为苯环结构,E2吸收带与B吸收带的吸收比(A1/A2)非常接近,只不过是双峰特征波长λ1、λ2各不相同;由于山梨酸K吸收带强度非常强和R吸收带强度相当弱,以致于二者比值相当大;而脱氢乙酸在λ1=230 nm与λ2=292 nm处的吸收比大约为2倍。杂质的存在都会使它们之间的比值发生偏离,吸收比的可靠性依赖于波长的选择及吸收值的区别,波长应尽量选择在具有最大吸收所对应的波长λmax处。因此,如果待测物与标准物质的最大吸收波长λ1和λ2都相同,及二者的双波长吸收比(A1/A2)接近,则可认为待测物与标准物质是同一物质。例如,图11待测物检出的糖精钠λ1、λ2及吸收比(A1/A2):13.4,与表1中标准物质的糖精钠λ1、λ2相同及吸收比(A1/A2):12~14相当接近。
4.3 色谱峰紫外归一化光谱图对比法
一般选保留时间处的色谱峰光谱图进行对比,如果待测物色谱峰的紫外光谱图与标准物质色谱峰的紫外光谱图是重合的——只是色谱峰的前沿、峰谷、峰顶、峰尾各点组成的光谱图只在振幅响应上有大小的差别,则可以判定,待测物与标准物是同一物质[17](如图10检出糖精钠);反之,如果二者的紫外光谱图是完全不一致,则二者就不是同一物质。如果待测物色谱峰光谱图的峰顶波长与标准物质色谱峰光谱图的峰顶波长(λmax)相同,但是待测物色谱峰光谱图其它位置出现不一致或是出现其它杂峰,这时就说明样品有杂质未能有效分离,色谱峰不纯,这时就得改进样品前处理,或是改变流动相配比,或是采用柱效更好的色谱柱,或是严格采用国家安全标准里的仲裁方法进行验证。由于液相色谱仪的紫外光度检测器波长存在示值误差(最大允许误差±2 nm[21]),应定期对液相进行检定或校准;加上紫外光度检测器使用过程中会发生漂移,因此对于标准物质和待测物色谱峰光谱图的峰谷、峰顶也是允许存在±2 nm误差(如图10检出的糖精钠在B吸收带λmax=263 nm,偏离2 nm)。

图10 蜂蜜话梅样品中检出糖精钠的紫外可见光谱图
5 结论
在选定的色谱条件下,应首先通过二极管阵列检测器对标准物质的色谱峰进行扫描,得到该标准物质的紫外可见光谱图,建立本机标准物质的紫外可见标准图谱库;当待测物色谱峰的保留时间不一致时或是出现可疑色谱峰时,采用本机的紫外可见标准图谱库对可疑待测物的进行定性分析是可行的和必要的;液相色谱法的定量检测和紫外光谱图定性鉴别同时进行是高效的,发现可疑待测物时可以不用再通过改变流动相等其它方法重复试验,可以节省试验时间,从而达到液相定性分析和定量分析的统一。
在实际检测应用中,通常采用色谱峰紫外归一化光谱图对比法就能够对待测物的定性分析做到不漏检、不误判,也可以轻松地剔除小杂质峰,该方法也是最为简便直观有效的。此外,带有定时间扫描的紫外检测器液相色谱仪也可以借鉴上述方法,根据色谱峰的保留时间用定时间程序来进行光谱扫描。因此,通过液相色谱法的紫外可见光谱图进行定性分析和简单分子结构推断的方法是可行的,建立本机乃至全国共享标准物质的紫外可见标准图谱具有重要的参考意义。
