衍生化-溶剂提取-气质联用法测定水中痕量烷基汞
2021-09-13杨丽莉王美飞胡恩宇戴维
杨丽莉 王美飞 胡恩宇 戴维
摘要:建立衍生化-溶剂提取-气质联用(GC-MS)测定水中痕量烷基汞的分析方法。用巯基固相萃取柱富集水中的烷基汞,4 mol/L 盐酸洗脱,在HAc-NaAc缓冲体系(pH 4~5)下与50μL 四丙基硼化钠(1% THF)衍生反应,0.5 mL 正己烷提取烷基汞的衍生物,采用气相色谱-质谱联用选择离子方式(SIM)进行检测,以4-溴氟苯为内标定量,在2~200 ng/L 范围内线性良好,1 L 水样中甲基汞和乙基汞的检出限可达1 ng/L 以下。实际样品添加回收率在69.7%~80.9%之间,准确度好;平行5次进行精密度试验,相对标准差在8.5%~10.3%之间。结果表明:方法稳定性好、操作方法实用,可用于水中痕量烷基汞的测定。
关键词:烷基汞;衍生化;溶剂提取;气相色谱-质谱联用;水样
中图分类号: O657.63;X832文献标志码: A文章编号:1674–5124(2021)12–0073–06
Determination of alkyl mercury in water by derivatization- solvent extraction-gas chromatography -mass spectrometry
YANG Lili,WANG Meifei,HU Enyu,DAI Wei
(Nanjing Environment Monitoring Center, Nanjing 210013, China)
Abstract: A sensitive and repeatable method was developed and applied to determine alkyl mercury in water byderivatization- solventextraction-gaschromatography-massspectrometry (GC-MS). The watersamples were extracted by SH solid-phase extraction column. The SPE column was eluted by 4 mol/L HCl and the acidic solution was adjusted to neutral. Then HAC-NaAC buffer (pH 4-5) and 50μL sodium tetrapropylborate (NaBPr4) solution (1% THF)were added to the solution. The derivatives can be completely extracted by 0.5 mL n-hexane. The analytes were determined by GC-MS in SIM mode and quantified by internal standard method. The results showed that the alkyl mercury derivatives had a good linear relationship in the rang 2 ng/L to 200 ng/L. The limits of detection were less than 1 ng/L in about 1 L water samples. The recoveries of the analytes in real water samples spiked with standards were all in the range of 69.7% to 80.9%. Th erelative standard deviations (RSD) were between 8.5% to 10.3%(n=5). The results show that the method is sensitive and accurate for the determination of the alkyl mercury in water samples.
Keywords : alkyl mercury; derivatization; solvent extraction; gas chromatography-mass spectrometry; water sample
0引言
環境八大公害事件之一的“水俣病”发生以来,世界各国达成了汞污染防治的共识,2017年《水俣公约》正式生效,我国也将和所有签约国一道严格控制汞排放和防治汞污染。汞(Hg)及其化合物在自然界中分布广泛,由于易与大部分金属形成合金,在传统和现代工业、化学药物和电子电器中都得到广泛应用,由此产生的废气、废水和废渣都会将汞污染转移至环境中,存在于环境中的汞有多种形态,可以在大气、水、土壤、生物体中迁移、循环和累积,是环境中广泛存在的污染物。汞的毒性与形态密切相关,与其他重金属不同在于可转化为毒性更强的有机汞(主要是甲基汞和乙基汞),兼有重金属和有机物双重毒性特点,可通过食物链放大最终毒害人体健康。特别是毒性最大的甲基汞,经肠道吸收后可使肝、肾、脑等多器官受到伤害,损伤神经系统,这也是“水俣病”的主要原因。水生生物特别是鱼类是人类摄取烷基汞的重要途径,因此对于水环境中汞的控制也有别于其他重金属,已根据毒性特点进行了形态控制,我国 GB 3838—2002《地表水环境质量标准》[1]、GB 5749—2006《生活饮用水卫生标准》[2]对总汞和烷基汞分别给出了评价限值,而烷基汞的标准限值更是达到了ng/L 水平。由于水中烷基汞浓度极低和仪器灵敏度的限制,水中烷基汞一直都是分析测试的难点,测定方法基本都是先富集再检测,富集材料大多选择了能与烷基汞产生鳌合作用的含巯基材料,有巯基纤维、巯基树脂、巯基葡聚糖凝胶、巯基活性炭、巯基键合硅胶等[3-5]。由于烷基汞兼有无机和有机物特性,检测技术则是从传统的气相色谱(GC-ECD )[6-7]发展到多技术联用设备,如气相色谱、高效液相色谱等分离设备与单级质谱、串联质谱、原子吸收光谱、原子荧光光谱、电感耦合等离子质谱等多种检测设备联用[8-15]。本文采用商品巯基固相萃取柱吸附水中烷基汞,盐酸洗脱,在缓冲体系中丙基衍生化反应,创新性地采用正己烷溶剂萃取衍生物,再用 GC/MS 选择离子模式检测,内标法定量,与原有国标相比,通过衍生化反应,可改善烷基汞的色谱行为,提高检测灵敏度;而与较为普及的气相色谱-冷原子荧光联用法相比,本方法条件比直接使用吹扫等方法容易控制,稳定性好,对处理好的样品可以重复进样分析,而且衍生物在提取溶剂中保存期长,可实现批量前处理和批量进样,解决了传统方法色谱行为和稳定性差的问题,灵敏度和准确度均能满足水质检测要求,经实际应用,效果良好,可应用于水环境中汞形态分析、风险评估和迁移转化等研究工作。
1实验部分
1.1仪器和试剂
仪器:7890B-5977A 气相色谱-质谱联用仪, EI 离子源(美国 Agilent 公司);HP -5MS 毛细管色谱柱(30 m ×0.25 mm ×0.25?m,美国 Agilent 公司)。
氯化甲基汞和氯化乙基汞标准溶液(1000 mg/L,Accustandard公司),用正己烷稀释成20.0 mg/L 的标准工作溶液,4℃冰箱密闭避光保存,备用。
四丙基硼酸钠(Accustandard公司)。将四丙基硼酸钠配成1%( V/ V)的四氢呋喃(THF)溶液。
内标4-溴氟苯(1000 mg/L , Chem Service 公司)。用正己烷稀释配制成50.0 mg/L 的标准溶液,备用。
用0.2 mol/L 乙酸和0.2 mol/L 的乙酸钠溶液配制 pH 值4~5的缓冲溶液。
Cleaner SH-SPE 固相萃取柱(500 mg/6 mL,天津艾杰尔科技有限公司)。
四氢呋喃和正己烷均为色谱纯试剂。实验用水为 Milli-Q 超纯水系统(美国 Millipore 公司)制备,其余均为市售分析纯级试剂。所用塑料和玻璃器皿均在1 mol/L 盐酸中浸泡24h 后洗净。
1.2气相色谱-质谱分析条件
GC 条件:柱温60℃保持1 min,以10℃/min 升至80℃,然后以25℃/min 升至200℃;进样口温度250℃。载气:高纯氦(99.999%),柱内流量采用恒流控制:1.0 mL/min。进样方式:分流进样,分流比5∶1,溶剂延迟时间2.5 min;进样量1.0?L。
MS 条件:传输线280℃,四极杆温度:150℃,电子轰击离子源(EI):70 eV,温度230℃。选择离子监测模式(SIM),甲基汞衍生物特征离子为 m/z 217,m/z 260和 m/z 202,定量离子为 m/z 217;乙基汞衍生物特征离子为 m/z 274,m/z 231和 m/z 202,定量离子为 m/z 274;内标4-溴氟苯特征监测离子为 m/z 174和 m/z 176,定量离子为 m/z 174。
1.3样品采集与保存
用聚乙烯或聚丙烯塑料瓶采集水样,样品采集后按照 GB 14204—1993的保存方法,加入盐酸调节 pH 在1~2,每升样品加入2 mL 饱和硫酸铜溶液作为抑菌固定剂,低温避光保存。
1.4样品处理
固相萃取柱预处理:将固相萃取柱用纯水浸润,再加入5 mL 4mol/L 盐酸浸泡20min,用纯水淋洗至中性,备用。
富集:取1 L 水样,调节 pH 为3~4,过滤去除悬浮物后连接固相萃取柱,调节真空压力,使得水样低于10 mL/min 通过固相萃取柱富集,抽干后,关闭真空泵。
洗脱:在萃取柱中加入2 mL 4 mol/L 盐酸,浸泡10 min 以上,用洗耳球挤出洗脱液,收集洗脱液于10 mL 聚乙烯离心管中,用 NaOH 溶液调节至近中性,再加入3 mL 乙酸-乙酸钠缓冲溶液( pH 值4~5),加入纯水将体积加至约9 mL,加入50?L 衍生化试剂、内标溶液和0.5 mL 正己烷,反应5 min 以上,振荡提取,将上层有机相部分转移至进样瓶中,进样1?L,如不能及时测定可密闭冷藏保存,15 d 内分析测试。
2结果与讨论
2.1分析条件优化
氯化甲基汞和氯化乙基汞經过丙基化反应后分别生成甲基丙基汞和乙基丙基汞,对衍生物进行扫描后确定定性定量离子,再通过调节气相色谱温度程序调节出峰时间,在5 min 内分离完全(见图1)。选择4-溴氟苯作为内标,在两个衍生物中间出峰,分离效果良好,色谱峰尖锐、对称。对衍生物进行质谱扫描,甲基汞和乙基汞衍生物的质谱图见图2,根据质谱信息,选择特征离子作为定量和定性离子。
2.2水中烷基汞 SPE 条件优选
由于巯基对汞的特效吸附性能,对于水中烷基汞的富集基本都是延续了这样的技术路线,有传统的巯基棉以及各种改良的巯基材料,为了保证富集材料的稳定性,选择了商品化巯基材料萃取柱,应用效果良好,保证了性能的稳定性。富集材料中的巯基对于多种金属元素都有着强吸附性能,将富集的烷基汞洗脱时应充分考虑其他金属离子的竞争问题,特别是作为保存剂加入的大量 Cu2+。本研究配置一定浓度烷基汞的水样通过巯基固相萃取柱,分别用3 mL 不同浓度的盐酸依次淋洗小柱,分别收集洗脱液,测定其中甲基汞( MeHg)和乙基汞(EtHg)的含量,评估不同酸度的洗脱效果,结果表明,按照 GB 14204—1993的洗脱方法并不能完全洗脱烷基汞,当盐酸浓度大于4 mol/L 后,3 mL 基本能全部洗脱。文献研究表明巯基材料对 Hg2+的吸附能力大于烷基汞和 Cu2+,可通过调节洗脱液酸度控制洗脱目标金属离子,但是由于样品保存时加入了大量的硫酸铜,在 Cu2+大量存在的情况下,加大盐酸浓度才能保证烷基汞的洗脱效率,实验现象也证实了这点,洗脱液呈现明显的含铜溶液的蓝绿色。
2.3衍生化试剂及衍生条件的选择
水中烷基汞常以氯化烷基汞的形式存在,盐酸洗脱后形态不变。氯化烷基汞的气相色谱行为和响应效果都不太好,存在系统吸附和比较严重的峰拖尾现象,为了解决这个问题,传统的 GC 分析方法在分析前先用含汞的溶液饱和色谱分离系统,但是效果并不稳定,后续发展的研究通过衍生化将氯化烷基汞转变成烷基汞,提高烷基汞的挥发性,减少吸附性,继而提高检测灵敏度[16-19]。常用的衍生化试剂有四乙基硼酸钠、四丙基硼酸钠和四苯基硼酸钠,本文对三种试剂的衍生化效果进行了比较,四乙基硼酸钠衍生反应快速、挥发性最好,是很多顶空、吹扫捕集等方法所选择的衍生方法,但是衍生试剂配制后稳定性差,必须马上使用,而且衍生反应迅速,衍生物在水相中不稳定;而四苯基硼酸钠由于苯基的位阻效应,衍生物反应速度较慢,而相比而言丙基化衍生物稳定性和挥发性是最佳的。虽然顶空或吹扫捕集的自动化程度高,顶空、吹扫等前处理设备无法控制衍生试剂的反应时间和在水相中的保持时间,加上传输管线等产生的吸附效应,实验结果显示稳定性比较差;另外由于在水中衍生后只能测定一次,一旦由于仪器不稳定或者连接出问题,样品必须重新进行富集等复杂的前处理过程,时间成本更大。本文通过实验选择四丙基硼酸钠进行衍生反应,根据衍生物特点采用溶液萃取的方式提取至有机相中,更利于测试样品的保存和集中分析测定。
2.3.1衍生化缓冲溶液 pH 值
用0.2 mol/L 乙酸和0.2 mol/L 乙酸钠溶液配制不同 pH 值的缓冲溶液,分别为3.8、4.2、4.6、5.0、5.4和5.8,试验不同 pH 下衍生化效果。在9 mL 纯水中加入氯化甲基汞和氯化乙基汞标准溶液10?g,加入缓冲溶液1 mL,加入四丙基硼化钠(1% THF溶液)50?L 和内标溶液,再加入0.5 mL 正己烷,反应5min 后充分振摇,静置分层,取上层有机相进样分析,考察不同 pH 条件下的衍生效果,见图3。结果显示在 pH 4~5之间反应效果稳定,和文献研究结果相近[19],因此确定反应条件为乙酸-乙酸钠缓冲溶液(pH 4~5)。
2.3.2衍生试剂添加量
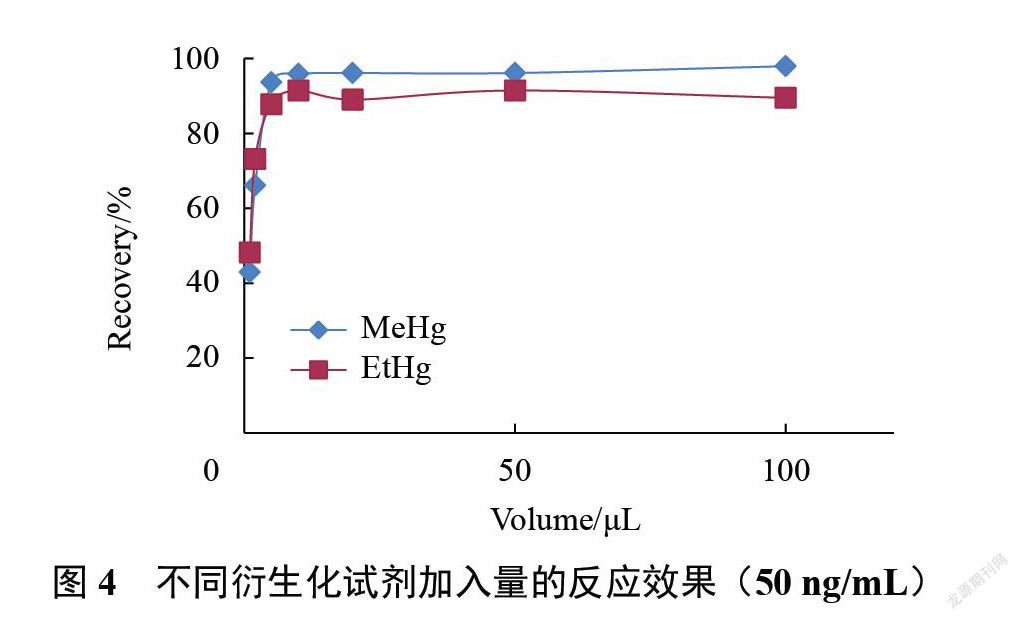
按照上述方法调节四丙基硼化钠溶液(1% THF溶液)的加入体积,考察衍生试剂量的影响,如图4所示,结果显示加入10?L 以后衍生效果基本保持稳定,为了保证衍生试剂过量或有其他物质消耗衍生试剂,实际样品分析时选择加入50?L 衍生试剂。
2.3.3反应时间
按上述方法加入50?L 四丙基硼化钠溶液,考察加入后不同时间衍生效果,结果显示衍生反应几乎是瞬间反应完全,2 min 以后效果基本保持稳定,实际操作时加入衍生试剂再加入正己烷后就可振荡萃取,基本不影响衍生效率。
2.3.4萃取溶剂和萃取次数
分别选择甲苯、正己烷、环己烷、乙酸乙酯作为萃取溶剂进行试验,结果表明,这几种溶剂对烷基汞衍生物的萃取效果差不多,最终选择毒性低的正己烷或环己烷作为萃取溶剂。用0.5 mL 正己烷萃取一次后将有机相全部吸取出来,再加入0.5 mL 正己烷萃取,对两次萃取液分别进样分析,结果显示一次萃取就能基本萃取完全。
2.3.5衍生物的稳定性
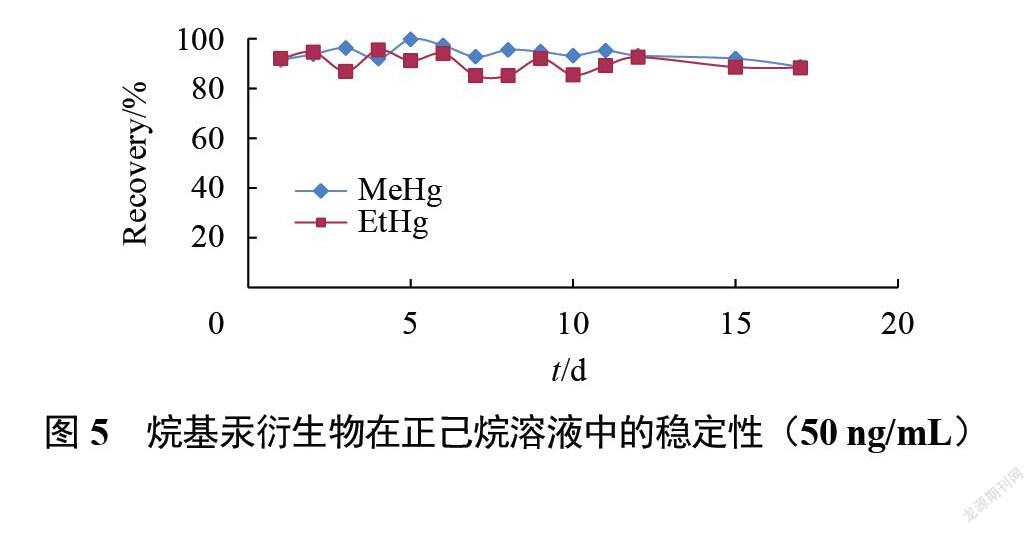
将萃取后含烷基汞衍生物的正己烷溶液分别于冷藏(4℃)和冷冻(–18℃)条件下保存在冰箱中,隔段时间进样分析,以最初烷基汞衍生物测定结果作为参比,考察其稳定性,冷藏保存结果见图5,结果显示衍生物在正己烷溶液中至少能保证15 d 不会发生分解等变化,冷藏和冷冻方式保存效果无显著性差异。
2.3.6容器材质的影响
由于汞吸附性比较强,在最初实验时,分析结果非常不稳定,查找各种原因后发现,玻璃材质实验器具对烷基汞的吸附较大而且不稳定,换成聚乙烯或聚丙烯塑料管以后衍生物效率稳定性较好,因此在整个实验过程中应尽量选择塑料材质的器具。此外在衍生化环节,将衍生化试剂和萃取溶剂一起加入,正己烷在上层可以起到封盖作用,对提高回收率有较好的作用。
2.3.7空白的控制
汞在环境中可说无处不在,而且具有较强的吸
附性,在操作环节使用的实验器具、耗材特别是巯基柱在开封、使用和保存过程中很容易出现汞空白较高的问题,虽然环境中大多污染干扰的是元素汞,不会在色谱分离时干扰本方法测定的烷基汞,因可被巯基材料富集,可能影响吸附效率,利用其吸附的可逆性,在进行水样吸附之前,先用4mol/L 盐酸进行淋洗净化,可去除吸附材料中吸附的汞,解决空白问题。对于实验中使用的离心管等可经泡酸、洗净以后用塑料密封保存备用。
2.4校准曲线和检出限
市售的烷基汞标准物质基本都是氯化烷基汞为主成分,本实验选择衍生工作曲线法进行绘制校准曲线,用与样品分析同批次试剂进行衍生化,可避免由于衍生条件的变化造成的系统误差。分别吸取不同体积的烷基汞标准溶液于9 mL 纯水中,加入量分别为2.0,5.0,10.0,20.0,50.0,100.0,200.0 ng,按照确定的实验方法进行衍生化和萃取,进样分析,以萃取液中理论浓度为横坐标,烷基汞衍生物定量离子峰面积与内标定量离子峰面积的比值为纵坐标,建立校准曲线,甲基汞和乙基汞衍生物线性回归方程分别为:y=0.0088x–0.296,相关系数0.9990;y=0.0048x–0.0271,相关系数0.9973,结果表明呈良好的线性关系。
在1 L 水样中加入5 ng 烷基汞标准物质,按照前述方法进行富集、衍生和测定,按3倍信噪比 ( S/N=3)计算,甲基汞和乙基汞的检出限分别为0.5 ng/L 和0.8 ng/L,能满足水质检测的要求。
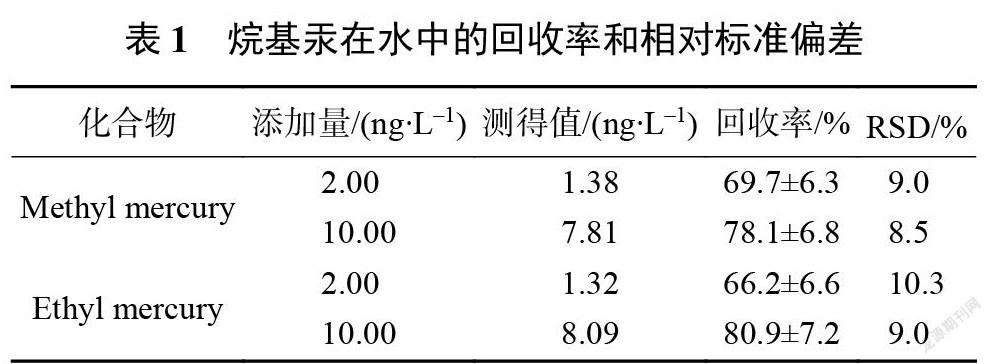
2.5回收率和精密度
选择实际地表水样品进行两个浓度水平的添加回收试验,计算相对标准偏差 RSD,评价精密度,结果见表1。结果显示,实际水样添加烷基汞的平均回收率在69.7%~80.9%之间,RSD 在8.5%~10.3%之间,该方法对水中痕量烷基汞的检测具有较好的精密度和准确度,可应用于水中烷基汞的测定。对照现行国家标准 GB/T 14204—1993,国标给出的加标浓度水平是5~400?g/L,远远高于方法的检出限10 ng/L 和20 ng/L,而地表水质量标准限值是1 ng/L,在痕量水平(2~20 ng/L)进行加标时,实际无法检出,在添加到100 ng/L 后,有较小的色谱响应,但是由于柱效不稳定,色谱峰拖尾严重,回收率在23.5%~45.3%之间波动,稳定性和准确度均不能达到较好的效果。
2.6方法应用
采用本方法对南京市饮用水源地的地表水样品(包括长江水样和湖泊水样)进行检测,全年共检测14个饮用水源地樣品,结果均为未检出,用 GB/T 14204—1993进行检测也是未检出,但是考虑到检出限的差异,本方法的未检出结果更具可信度,另外在实际应用中衍生物在正己烷中可长期保存的优点得到明显显现,稳定性更好,有利于批量分析测定。
3 结束语
本文建立了固相萃取衍生化-溶剂提取-气质联用法测定水中甲基汞和乙基汞的分析方法,经实际样品验证和与现行国标法比较,表明方法灵敏度高、精密度和准确度好,衍生物稳定,保存期较长,能满足地表水等水中烷基汞痕量检测的要求。
参考文献
[1]地表水环境质量标准: GB 3838—2002[S].北京:中国标准出版社, 2002.
[2]生活饮用水卫生标准: GB 5749—2006[S].北京:中国标准出版社, 2007.
[3]王启超, 汪淑哲.巯基葡聚糖凝胶的制备及其对微量重金属离子吸附性能的研究[J].化学试剂, 1985, 7(3):166-168.
[4]屈文, 苏继新, 曹丛华, 等.巯基键合硅胶的制备及其对贵金属离子的吸附性能[J].贵金属, 2000, 31(2):15-18.
[5]张伟安, 汪玉庭, 杨智宽.巯基壳聚糖对重金属离子的配合吸附性能[J].化学试剂, 2006, 28(2):65-67.
[6]环境甲基汞的测定气相色谱法: GB/T 17132—1997[S].北京:中国标准出版社, 1997.
[7]水质烷基汞的测定气相色谱法: GB/T 14204—1993[S].北京:中国标准出版社, 1993.
[8]赵云芝, 钱蜀, 王俊伟, 等.高效液相色谱-原子荧光联用测定天然水中烷基汞[J].中国环境监测, 2011,27(3):20-22.
[9]河滨, 江桂斌.固相微萃取毛细管气相色谱-原子吸收联用测定农田土壤中的甲基汞和乙基汞[J].岩矿测试, 1999,18(4):259-262.
[10]蒋红梅, 冯新斌, 梁琏, 等.蒸馏-乙基化 GC-CVAFS 法測定天然水体中的甲基汞[J].中国环境科学, 2004(5):568-571.
[11]王琳, 耿勇超, 董铮, 等. HPLC-ICP/MS联用测定废水中甲基汞和无机汞[J].环境监测管理与技术, 2010, 22(4):44-46.
[12]刘静, 王俊伟.固相萃取/液相色谱-原子荧光法测定地表水中的甲基汞[J].四川环境, 2017, 36(5):24-28.
[13] ZHENG H, HONG J J, LUO X L. Combination of sequential cloudpointextractionandhydridegenerationatomic fluorescencespectrometryforpreconcentrationand determinationofinorganicandmethylmercuryinwater samples[J]. Microchemical Journal, 2019, 145:806-812.
[14] XIE S S, GAN W E, HUANG M. Determination of methylmercury inwatersamples with electromagnetic inductionthermal desorption/pyrolysis coupled to atomicfluorescencespectrometry[J]. International Journalof EnvironmentalAnalyticalChemistry, 2014, 94(11):1150-1160.
[15] T?RKERBAR,?ABUKD,YAL?INKAYAO. Preconcentration, speciation, and determination of mercury by solidphaseextractionwithcoldvaporatomicabsorptionspectrometry[J]. Analytical Letters, 2013, 46:1155-1170.
[16]吴建刚, 赵金平, 赵志南.四丙基硼化钠衍生吹扫捕集冷原子荧光光谱法同时测定水中的甲基汞和乙基汞[J].环境化学, 2015, 34(2):390-391.
[17] SHARIF A, MONPERRUS M, E. Determinationof methyl mercury and inorganic mercury in natural waters at the pgL-1 level: Intercomparison between PT-GC-Pyr-AFS andGC-ICP- MSusingEthylationorPropylationderivatization[C]//E3S Web of Conferences, 2013.
[18] CENTINEO G, GONZ?LEZ E B, SANZ-MEDEL A, et al. Multielementalspeciationanalysisoforganometallic compounds ofmercury, lead and tin in natural water samples byheadspace-solidphasemicroextractionfollowedbygas –mchromatographyass spectrometry[J].Journal of Chromatography A, 2004, 1034:191-197.
[19] MONPERRUSM,TESSIER E. Simultaneousspeciationof mercury and butyltin compounds in naturalwaters and snow by propylationandspecies-specificisotopedilutionmass spectrometry analysis[J]. Anal Bioanal Chem, 2005, 381:854-862.
(编辑:莫婕)
