超高效液相色谱-串联质谱法检测中药饮片中8种黄色素类染色剂*
2021-09-12刘延忠刘东升朱旭江姚世霞
刘延忠,刘东升,朱旭江,姚世霞
(1.甘肃省医疗器械检验检测所,甘肃 兰州730070;2.甘肃省药品检验研究院,甘肃 兰州730070)
中药饮片作为中药提取物和中成药的基本原料,市场需求量大,染色剂染色伪制的常用中药饮片的外观性状接近正品,不易辨别,导致不法分子将假、劣药材染色后冒充正品[1-2]。据报道,大部分黄色类染色剂不利于人体健康,如金胺O、金橙Ⅱ等黄色素类染色剂对人体可造成不同程度危害,故禁止用于中药饮片的染色。近年来,虽然药监部门加大了中药非法染色的打击力度,金胺O等染色剂的染色现象有所减少,但仍有不法商贩使用其他黄色素类染色剂进行非法染色。目前,国家药品监督管理局已颁布的中药饮片染色掺假物质补充检验多采用薄层色谱(TLC)法、高效液相色谱(HPLC)法对单个药材及饮片中的一种或几种色素进行检测[3-4],品种覆盖面窄,检测方法灵敏度低,准确性差,易出现假阳性,不能有效控制中药饮片的质量。超高效液相色谱-串联质谱联用法(UPLC-MS/MS)广泛用于中药非法染色剂的检测[5-14]。本研究中建立了同时测定蒲黄、黄芪、黄连、黄柏和延胡索等多种中药饮片中8种黄色素类染色剂含量的UPLC-MS/MS法,为中药饮片中黄色素类染色剂的检测提供参考。现报道如下。
1 仪器与试药
1.1 仪器
LCMS-8045型超高效液相色谱-三重四极杆液质联用仪(日本岛津公司);ME204型和MS205DU型分析天平(瑞士梅特勒-托利多公司,精度均为0.01 mg);KQ-500DE型数控超声波清洗器(昆山市超声仪器有限公司,功率为500 W,频率为40 kHz);XK80-A型快速混匀器(江苏新康医疗器械有限公司);SHY-2A型多功能水浴恒温振荡器(金坛市大地自动化仪器厂);5430R型离心机(德国艾本德公司);HGC-24A型氮气吹干仪(天津市高科技发展有限公司);Research plus加样枪(德国艾本德公司,规格为10,100 μL);Xiboshi一次性针式过滤器(美国戴安公司,规格为0.22,0.45 μm)。
1.2 试药
柠檬黄对照品(批号为30117,纯度为90.0%);日落黄对照品(批号为91001,纯度为87.0%),金胺O对照品(批号为00701,纯度为79.0%),金橙Ⅱ对照品(批号为21116,纯度为96.3%),碱性橙22对照品(批号为6A16a050,纯 度 为91.5%),均 购 于Dr.Ehrenstorfer GmbH;金橙Ⅰ对照品(上海源叶生物科技有限公司,批号为S11N8D48136,纯度≥98%);灿烂黄对照品(批号为A2689A,纯度为95%),皂黄对照品(批号为D1714050,纯度为98%),均购于上海Aladdin公司;乙腈、甲醇、乙酸铵、甲酸铵均为色谱纯,其他试剂均为分析纯,水为超纯水。药材样品信息见表1。

表1 药材样品信息Tab.1 Information of Chinese herbal pieces
2 方法与结果
2.1 UPLC-MS/MS条件
色谱条件:色谱柱为Thermo AcclaimTMRSLC 120 C18柱(100 mm×2.1 mm,2.2 μm);流动相为乙腈(A)-0.02 mol/L乙酸铵溶液(B),梯度洗脱(0~12 min时90%B→55%B,12~20 min时55%B→30%B,20~40 min,时30%B→15%B);流速为0.2 mL/min;柱温为30℃;进样量为5 μL。
质谱条件:电离方式为正离子电喷雾离子源(ESI+),反应监测模式(MRM);DL管温度为350℃;干燥气流速为10 L/min;雾化气流速为3.0 L/min;接口电压为4.0 kV。总离子流图见图1,各成分质谱信息见表2。

表2 8种黄色表类染色剂质谱信息Tab.2 Mass spectrum information of eigh yellow pigments

图1 8种黄色素类染色剂混合对照(中等线性浓度)的总离子流图1.Lemon yellow 2.Sunset yellow 3.Brilliant yellow 4.OrangeⅠ5.Golden orangeⅡ6.Auramine O 7.Soap yellow 8.Alkaline orange 22Fig.1 Total ion flow diagram of mixed reference(medium linear concentration)of eight yellow pigments agents
2.2 溶液制备
分别取柠檬黄、日落黄、金橙Ⅰ、灿烂黄、金胺O、金橙Ⅱ、皂黄和碱性橙22对照品各10 mg,精密称定,加70%乙醇分别制成质量浓度为100 μg/mL的单标混合贮备液,于4℃条件下保存。取上述单标混合贮备液各1~3 mL,置50 mL容量瓶中,加70%乙醇定容,摇匀,即得混合对照品溶液。取样品适量,粉碎,取2.0 g,精密称定,加70%乙醇20 mL,超声处理(功率为250 W,频率为40 kHz)30 min,冷却至室温,用70%乙醇补足减失的质量,摇匀,取续滤液,即得供试品溶液。
2.3 方法学考察
线性关系考察和检测限确定:分别精密吸取2.2项下稀释1倍后的单标混合贮备液,加70%乙醇,分别制成系列混合对照品溶液。注入超高效液相色谱-三重四极杆液质联用仪,按2.1项下色谱条件测定染色剂的峰面积积分值。以混合对照品溶液质量浓度为横坐标(X,μg/mL)、峰面积为纵坐标(Y)绘制标准曲线,得到近原点的一条直线,计算回归方程和相关系数。结果见表3。可见,8种黄色类染色剂质量浓度在0.2~30.0 μg/mL范围内与峰面积线性关系良好。方法的检测限为0.05~0.10 μg/g。详见表3。
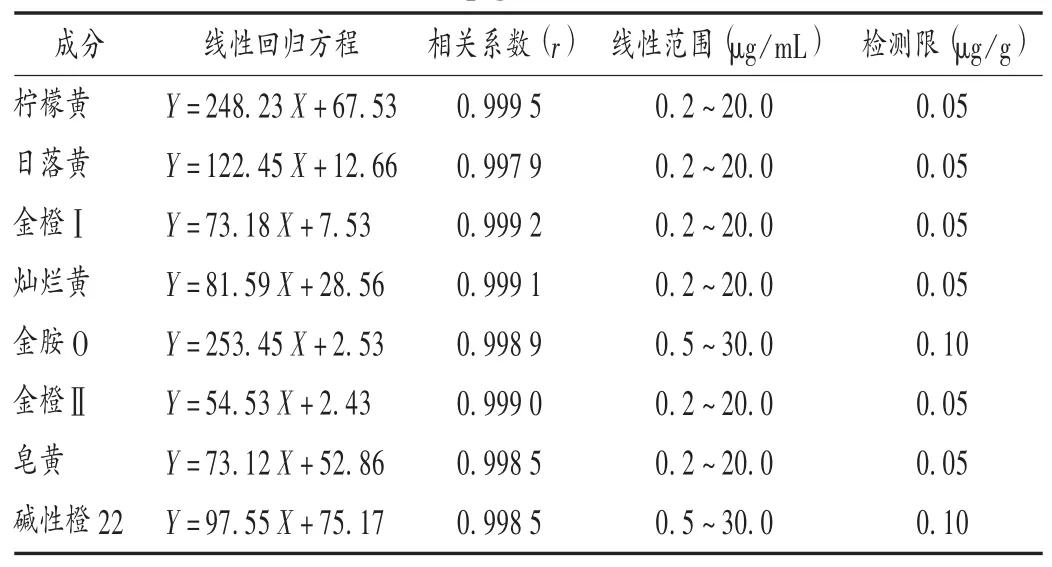
表3 8种黄色素类染色剂回归方程、线性范围和检测限Tab.3 Regression equation,linear range and LOD of eight yellow pigments
精密度和准确度试验:采用标准添加法,称取完全不含染色剂的空白样品6份,每份1.0 g,精密称定,分别精密加入混合对照品溶液各2 mL,按2.2项下方法制备供试品溶液,按2.1项下色谱条件进样测定。结果平均回收率为89.83%~99.52%,RSD为2.14%~3.67%(n=6)。详见表4。
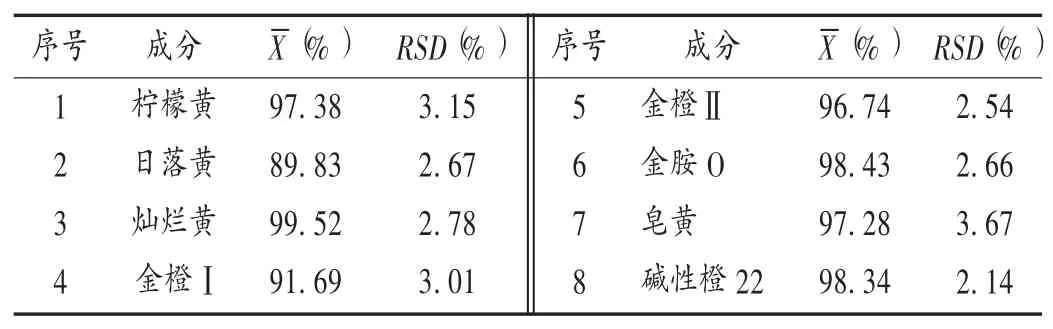
表4 精密度和准确度试验结果(n=6)Tab.4 Results of precision and accuracy tests(n=6)
2.4 样品中非法添加黄色素类染色剂检测
采用UPLC-MS/MS法,按2.1项下色谱条件对26批蒲黄(2批)、黄芪、黄连、黄柏和延胡索(各6批)样品中非法添加黄色素类染色剂进行检测,结果样品均未检出黄色素类染色剂。
3 讨论
3.1 提取条件优化
曾考察不同提取溶液(甲醇、乙醇、70%甲醇溶液、70%乙醇溶液)对测定结果的影响,结果经70%乙醇溶液提取,供试品溶液进样后色谱峰峰形良好,且杂峰较少。综合考虑,以70%乙醇溶液为提取溶剂。考察不同提取方法(浸泡、超声、回流提取法)对测定结果的影响,结果超声提取法简便、快捷,且提取效率较高,故采用超声提取法。考察不同体积(10,20,30,50 mL)的70%乙醇溶液和不同超声时间(15,30,45,60 min)对测定结果的影响,最终选用20 mL 70%乙醇溶液超声处理30 min。
3.2 质谱条件优化
根据8种染色剂分子的结构特征,分别在正离子和负离子模式下进行全扫描,结果在正离子模式下各染色剂的响应值均高于负离子模式下的响应值,故选用正离子模式;通过UPLC的自动进样器直接进样(不连接色谱柱),对毛细管电压、锥孔电压及质谱分辨率等条件进行优化,确定接口电压为4.0 kV。在拟订质谱条件下,分别以分子离子为母离子,对其子离子进行全扫描,选取响应最高且干扰较小的子离子作为定量离子(MRM模式),以响应值次之的作为定性离子,并对目标化合物二级质谱的碰撞能量等质谱分析参数进行优化。最终确定了质谱条件。
3.3 流动相优化
在质谱分析过程中,由于待测样品(阳性含染色剂)中含有偶氮键和季铵盐,故流动相中常加入一定浓度的甲酸和铵盐(甲酸铵或乙酸铵),有助于正离子的响应和峰形的改善。选择乙腈-0.02 mol/L乙酸铵溶液、甲醇-0.02 mol/L乙酸铵溶液、乙腈-0.02 mol/L甲酸铵溶液和甲醇-0.02 mol/L甲酸铵溶液4种流动相体系进行分析。结果显示,采用乙腈-乙酸铵体系进行梯度洗脱时,样品分离度和峰形均较好,且能进一步提高分析样品的信噪比和灵敏度。
3.4 方法评价
本研究中建立了同时测定中药饮片中可能添加的8种黄色素类染色剂的UPLC-MS/MS法,该方法稳定性好,可操作性强,准确度高,提高了工作效率,降低了检测成本。与HPLC法相比,进一步提高了方法的准确度和灵敏度,检测的色素种类较全面,有效地提高了检测速度,可为监测中药饮片中非法染色提供参考。
