潜在的胚胎干细胞自我更新与多能性的调控基因的鉴定:基于随机森林算法
2021-09-10曾彭归航唐秀晓吴庭芩李茫茫丁俊军
曾彭归航,唐秀晓,吴庭芩,田 奇,李茫茫,丁俊军
1南方医科大学基础医学院,广东 广州 510515;2中山大学中山医学院,广东 广州510080
小鼠胚胎干细胞(mESCs)来自早期胚胎内细胞团并可在体外进行无限期的培养,具有自我更新和分化为所有成体细胞的能力,这些特性使得mESCs适于构建疾病模型和应用于再生医学的研究中[1]。深入解析干细胞自我更新和多能性调控的分子机制是干细胞应用的基础。其中,系统鉴定与干细胞自我更新和多能性相关的基因是一个重要的方向。
目前虽然已经有一些研究报导了大规模鉴定这些基因的方法,其中包括基于实验的功能研究和基于计算的研究,但都存在一定的局限性。如,基于实验的功能研究主要是通过RNA干扰来筛选基因[2-5],这些研究较为耗时费力,难以覆盖全基因组上的基因,且由于文库和报告系统的不同,这些研究获得的结果彼此之间重合率较低。为解决这些问题,一些基于机器学习的计算方法被提出,如基于支持向量机(SVM)和由46个基因组成的阳性集,使用转录因子的染色质免疫共沉淀测序(ChIP-seq)和mRNA微阵列信息来预测干细胞多能性相关基因[6];基于遗传算法和SVM使用mRNA微阵列信息来寻找mESCs的生物标志物[7];基于SVM使用蛋白质互作信息来预测多能性相关的蛋白质[8],均能实现全基因组水平的筛选。但是,这些研究仅仅使用了少量类型的数据如基因表达数据和转录因子结合数据,同时仅使用了较小的阳性集。基于蛋白质互作信息来构建模型将使得结果仅限于蛋白质编码的基因,而忽略了非编码的基因,如microRNA和长非编码RNA,可能会遗漏掉一些重要的自我更新与多能性相关基因。
近年来,许多关于mESCs的多组学数据出现了,包括组蛋白修饰、染色质可及性、转录因子及结构蛋白在染色质上的结合等,研究表明这些多组学数据与干细胞自我更新及多能性相关基因的调控有关[9-17],因此根据基因的这些多组学特征可能可以预测出新的自我更新及多能性基因。目前还没有研究统一整合这些数据来进行预测,因此本研究构建了基于随机森林算法的模型,使用了10倍于前述研究大小的阳性集,整合这些多组学数据来鉴定潜在的干细胞自我更新及多能性调控基因。
1 材料和方法
1.1 数据的准备及可视化
本文中的多组学二代测序数据由多个mESCs相关的研究中收集而来。从EMBL-EBI数据库[18]获取原始测序数据后,对所有测序数据首先使用Trim Galore软件进行质量控制,随后比对到mm9 基因组(Ensembl release 67)。RNA-seq数据由STAR 软件进行比对,用HTSeq-count 对每个基因上唯一比对的序列进行计数[19],并进一步转化为标准化的表达值,即基因的每千碱基的转录每百万映射读取的读段(RPKM)。ChIP-seq及利用转座酶研究染色质可及性的高通量测序(ATAC-seq)数据使用bowtie2软件进行比对[20]获得比对结果bam文件,后续使用bedtools[21]根据bam文件计算特定区域内的读段数量。bam文件由deepTools[22]转化为bigwig文件并在WashU[23]网站进行可视化。本文中使用的测序数据相关 的GEO 数据库编号为:GSE29218[24],GSE99518[9],GSE22562[25],GSE106176[26],GSE43231[27],GSE112808[28],GSE11431[29],GSE96107[30],GSE80820[31]。
1.2 基因的染色质特征的比较
对于全基因组范围内的每个基因(不包括线粒体基因),我们以RPKM来表征其表达值,其它多组学染色质特征则由ChIP-seq和ATAC-seq数据在启动子区域的读段的数目来表征。由于并非所有基因的启动子位置都已知,我们分别设定基因启动子为转录起始位点的上下游n=250/500/1000/3000 碱基的区域并计算读段数目,在比较基因相关多组学信号的高低时使用n=3000的条件,在后续模型的训练过程中这4个条件对应的数值都被输入以增加数据的维度。对于具有多个不同转录本及转录起始位点的基因,我们仅选择上下游3000碱基范围内RNA聚合酶II(RNAPII)信号最高的转录起始位点来进行计算。使用rstatix比较从ESCAPE数据库[32]收集的已被验证的自我更新与多能性相关基因(本文中简称为ESCAPE基因)与全基因组基因或随机挑选的相同数量的基因的RPKM 值、ChIP-seq 和ATAC-seq读段数目并使用ggplot2绘图。使用numpy进行数据处理并计算不同特征之间的皮尔森相关系数并用pheatmap绘图。
1.3 模型的选择
机器学习技术可以通过使用大量数据,对复杂系统的输入与输出之间的关系进行建模[33]。机器学习分为监督学习、无监督学习、半监督学习和强化学习等,已经被广泛用于整合生物系统中的各类数据以处理回归、聚类、降维和关联研究等问题,常用的算法分类为特征连接、贝叶斯、集成学习、多核学习等[34]。本研究中通过已有的特征来训练模型并预测结果属于监督学习的范畴,首先选择了常用的一些机器学习算法来构建模型并进行评估,包括:属于特征连接的SVM,属于贝叶斯的朴素贝叶斯网络,属于集成学习的随机森林和LGBM(LightGBM),属于多模型深度学习的神经网络。
1.4 模型的训练及评估
模型的训练需要特征矩阵、阳性集和阴性集。在本研究中,特征矩阵包含表征全基因组上每个基因的表达值以及其启动子区域上的其它多组学数据信息的数值。阳性集包括ESCAPE基因集中RPKM值大于1的基因,ESCAPE基因共计480个,经RPKM值筛选后剩余308个。阴性集包括全基因组范围内随机挑选的与阳性集相同数量的基因(n=308)。接着将输入数据等分为3份,其中1份作为测试集(n=205),其余的作为训练集(n=411)。使用scikit-learn[35]和训练集来构建模型并进行5折的交叉验证,最终使用测试集来进行独立测试。重复整个过程100次并记录交叉验证和独立测试的评估结果,使用ggplot2绘制箱线图展示AUC值。相关代码存储于:https://github.com/Devalock/Self-renewal。
1.5 功能富集分析
使用g:profiler[36]进行功能富集分析,Vennerable绘制韦恩图,ggplot2绘制排名最高的GO、KEGG条目及相关的FDR值。
1.6 细胞培养
mESCs细胞株R1(来自实验室细胞库),培养于明胶(BD)铺底的培养皿,使用包括15%血清,0.1 mmol/L β-巯基乙醇(Sigma),2 mmol/L L-谷氨酰胺(Thermo Fisher),0.1 mmol/L非必需氨基酸(Thermo Fisher),1%核苷混合物(Sigma),1.5%重组LIF(实验室制备)及50 U/mL 青霉素/链霉素(Hyclone)的DMEM 培养基(Hyclone)。
1.7 敲低细胞系构建
质粒构建:本研究中用于敲低Cct6a的短发夹RNA(shRNA)载体为pLKO-puro-shCct6a。所使用的pLKO载体(来自实验室质粒库)由pLKO.1修改而来[37]。从遗传扰动平台(https://portals.broadinstitute.org/gpp/public/gene/search)选择了两个已被验证的shRNA序列,序列如表1所示。
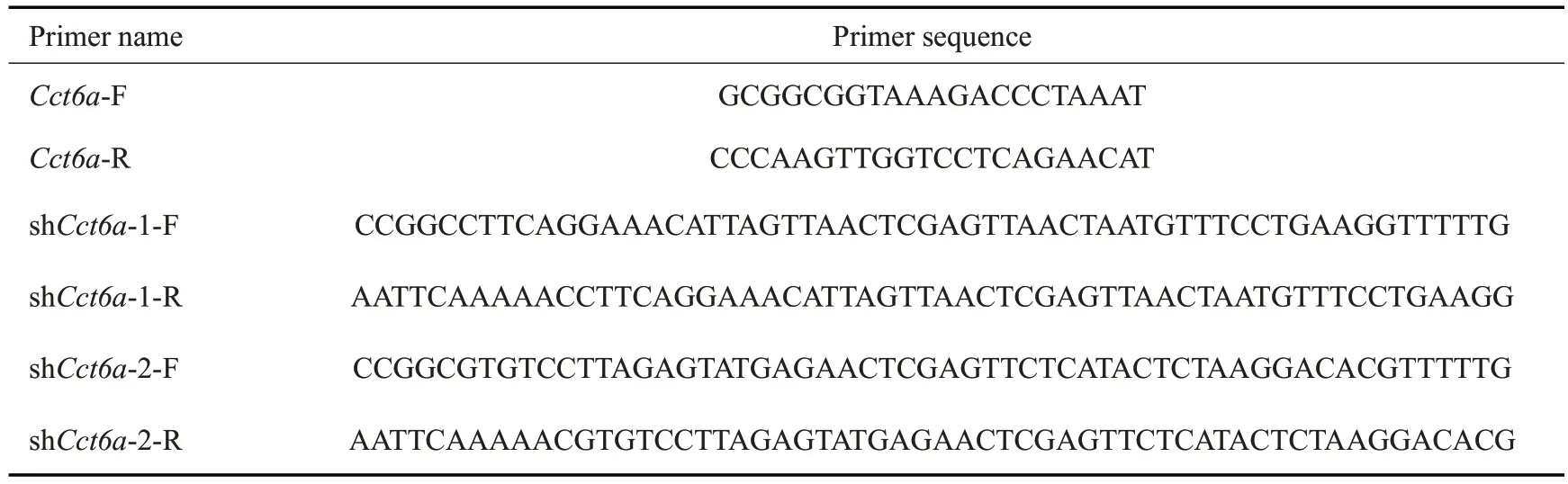
表1 所使用引物的序列Tab.1 Sequences of used primers
病毒包装:将16.76 μg目的质粒,11.26 μg pspAX2与7.33 μg vsvg(来自实验室质粒库)加到453 μL 灭菌水中,之后加入400 μL 2 mol/L CaCl2。取1.5 mL EP管,加入400 μL 2×BES(碧云天,磷酸钙法细胞转染试剂盒),用移液管在BES中吹打气泡,逐滴加入DNA混合物,室温孵育15 min。将提前24 h传代并接种到10 cm培养皿的293T细胞(来自实验室细胞库)的培养基更换为含25 nmol/L氯喹(APExBio)的新鲜的DMEM+10%FBS(Lonsera)培养基,并逐滴加入前述的混合物。24 h后更换培养基为不含LIF的mESC培养基,分别在48 h和72 h后收集病毒液,4000 r/min离心5 min,将上清转移置于4 ℃保存待用。
病毒感染及药物筛选:使用6孔板进行实验,病毒液与培养基比例为1∶3,加入终浓度为10 μg/mL 的polybrene(碧云天)。每个shRNA感染2孔,每孔5万细胞。24 h后使用终浓度为1 μg/mL的嘌呤霉素(索莱宝)进行药物筛选,48 h后使用终浓度为2 μg/mL的嘌呤霉素进行药物筛选。96 h后开展后续实验。
1.8 细胞活性测定
取1.5万细胞用3 mL mESC培养基重悬,取100 μL(500细胞/孔)的细胞悬液接种到明胶包被的96孔板上,周围1圈加100~200 μL PBS(Hyclone)。使用细胞计数试剂盒8(CCK8,同仁)每隔24 h添加10 μL的CCK8试剂孵育3 h后,用酶标仪检测吸光度A450nm。
1.9 克隆形成测定
取前述200 μL细胞悬液接种于6孔板的1孔中,培养5 d后使用碱性磷酸酶试剂盒(翊圣)染色并计数。
1.10 细胞周期分析
取前述剩余细胞的一半传代至6 孔板中继续培养。后收细胞(106左右),用PBS清洗2次,70%乙醇(广州化学试剂厂)重悬细胞,4 ℃固定过夜。300g离心3 min去除上清,用PBS清洗2次。配置染色剂,每1 mL的PBS 里含终浓度为3 μmol/L 的DAPI(Sigma)和终浓度为0.1%的Triton X-100(Amresco),每个样品加入300 μL染色剂,室温避光孵育30 min。使用流式细胞分析仪(CytoFLEX)进行实验数据的收集。用FlowJo软件对实验数据进行处理和分析。
1.11 实时定量PCR(qRT-PCR)
收集剩余细胞,首先使用Eastep Super 总RNA提取试剂盒(Promega)提取总RNA。使用反转录试剂盒将RNA反转录合成cDNA,将得到的cDNA产物稀释至2.5 ng/μL 作为模板,以GAPDH 作为内参来进行qRT-PCR检测基因的表达。将cDNA与ddH2O按3∶10的比例配置cDNA mix。引物序列包含在表1中,将引物稀释至10 mmol/L,上下游引物1∶1混合,将引物与qRT-PCR mix(Takara)按2∶15比例配置primer mix。用电子枪按照设计的qRT-PCR 模板将5.2 μL 的cDNA mix点入qRT-PCR板相应的孔中,点完后用保鲜膜覆盖qRT-PCR板的孔,2000g离心2 min;接着点6.8 μL的Primer mix,点完后贴qRT-PCR膜,2000g离心2 min;按qRT-PCR mix 对应的说明书设置qRT-PCR 仪,将qRT-PCR板放入仪器中,运行程序。反应结束后,根据软件Bio-Rad CFX Manager分析处理数据。
1.12 统计学分析
两样本间的比较使用Student'st-tests,细胞活性的比较使用Two-way analysis of variance(ANOVA)。数据结果以均数±标准差来表示,每个实验均重复3次,P<0.05为差异有统计学意义。
2 结果
2.1 已知的自我更新与多能性基因的染色质特征
在mESCs中已知的自我更新与多能性基因通常表现出特定的特征,包括较高的RNA-seq信号,激活性信号H3K27ac的富集,较高的染色质可及性以及转录因子的结合信号等;分化相关基因通常表现出相反的模式,如较低的RNA-seq信号,抑制性信号H3K27me3的富集,较低的染色质可及性等(图1A)。
ESCAPE基因是已经被实验验证的与自我更新或多能性相关的基因,统计结果表明相较于全基因组所有基因或随机挑选的相同数目的基因,ESCAPE基因的激活性信号显著更高而抑制性信号显著更低(图1B)。ESCAPE基因和全基因组范围上染色质特征之间的相关性分析(图1C)发现某些信号之间具有较高的相关性从而形成集群,如OCT4、SOX2、NANOG及ATAC-seq信号形成集群,CTCF与黏连蛋白SMC1和SMC3以及组蛋白第3亚基4号赖氨酸的单甲基化(H3K4me1)形成集群,RNAPII和H3K27ac的信号具有很高相关性;ESCAPE基因的信号模式与全基因组上的信号模式具有差异,不同集群之间的相关性更弱,H3K27me3与其他信号之间的相关性更弱,同时在全基因组范围内RNA-seq 的信号与其它信号没有正相关性,但在ESCAPE基因上与RNAPII的信号正相关。

图1 ESCAPE基因与其他基因的染色质特征具有差异Fig.1 ESCAPE genes differ from other genes in chromatin features.A:Genomic profiling of Oct4, Sox2, Nanog and Sox9.B:Comparisons of chromatin features between ESCAPE genes,all genes and randomly chosen genes;C:Pearson's correlation coefficients between chromatin features of all genes(left)and ESCAPE genes(right)(****P<0.0001,***P<0.001,**P<0.01).
2.2 使用机器学习方法训练并评估模型
使用多种机器学习分类器来构建模型并通过受试者特征曲线(ROC)的曲线下面积(AUC)进行评估,不同机器学习方法在交叉验证(图2A)与独立测试(图2B)的结果显示随机森林方法的AUC最高。拆分数据以单个特征进行训练,交叉验证(图2C)与独立测试(图2D)的结果发现整合使用所有特征可获得最好的效果,RNA-seq,RNAPII,YY1与H3K4me3信号对于模型贡献较高。具体的评估指标如表2、3所示,结果表明随机森林的表现最优,使用所有特征的AUC略高于单独使用RNA-seq 数据,在召回率(Recall),准确性(ACC),Matthews相关系数(MCC)等指标上有较大提升。基于以上结果整合所有特征并使用随机森林构建模型进行潜在目标基因的预测。

表2 不同分类器的交叉验证及独立测试(括号内)的评估指标Tab.2 Performance measures of cross validations and independent tests(in brackets)among different classifiers
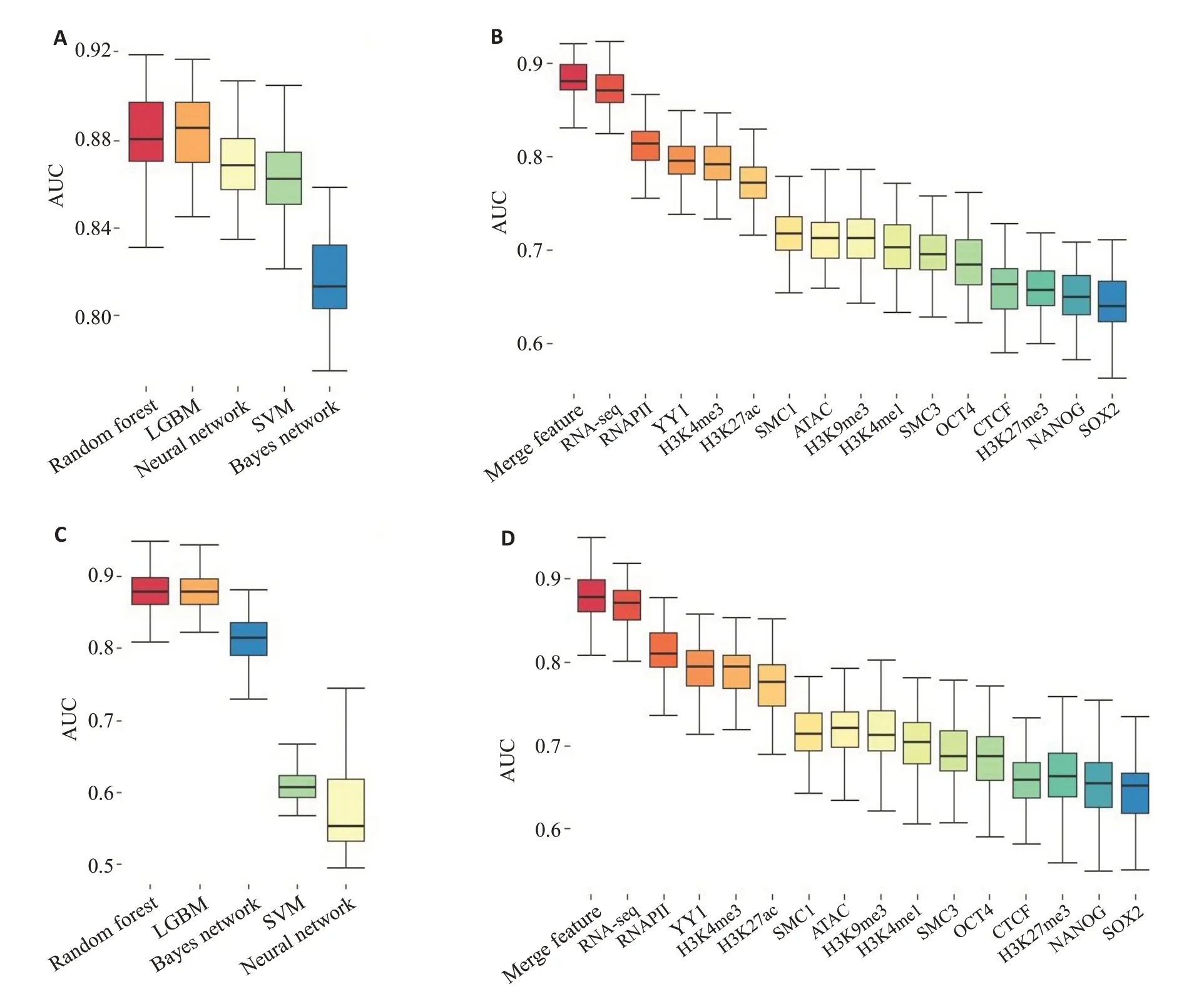
图2 使用随机森林并整合所有特征的模型具有最高的AUCFig.2 The model based on random forest with merged features has the highest AUC.A:Cross validations using different classifiers.B:Independent tests using different classifiers.C:Cross validations using different features.D:Independent tests using different features.
2.3 基于文献及实验验证预测的基因
基于2.2部分建立的模型对全基因组范围每一个基因进行打分,将模型的准确性达到最高时的预测分数0.85作为阈值,获得了893个潜在的目标基因(不包括ESCAPE基因)。对ESCAPE基因和潜在目标基因作GO(图3A)和KEGG富集分析(图3B),结果具有很高的重合度。
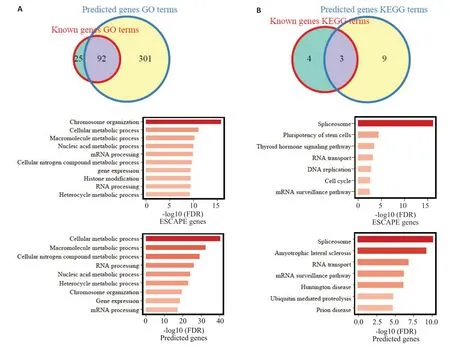
图3 ESCAPE基因和预测基因的功能注释具有很高的重合度Fig.3 Functional annotations of ESCAPE genes and the predicted genes have substantial overlap.A:Overlap of GO terms and the highest ranked GO terms.B:Overlap of KEGG terms and the highest ranked KEGG terms.
Cct6a基因的分数排名较高(表4)且没有文献提到其与自我更新或多能性相关,因此我们选取其作为功能验证的对象。使用shRNA在mESC中对Cct6a进行敲低,效率达到约80%(图4A),Cct6a 的表达显著下调(shCct6a-1组,P=0.0026;shCct6a-2组,P=0.0045)。使用CCK8计算细胞活力,结果表明敲低Cct6a的细胞增殖能力严重受损(P<2e-16,图4B),细胞的形态也发生较大改变(图4C)。细胞克隆形成实验表明大部分细胞死亡(shCct6a-1组,P=0.0053;shCct6a-2组,P=0.0049)(图4D),剩余的细胞也难以被碱性磷酸酶所染色(图4E)。使用流式细胞术计数处于细胞周期不同时期的细胞,处于G1 期的细胞增多(shCct6a-1 组,P=0.00038;shCct6a-2组,P=0.0037),处于S期的细胞减少(shCct6a-1组,P=0.021;shCct6a-2组,P=0.026,图4F)。
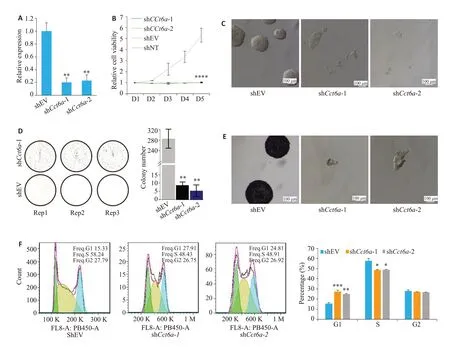
图4 敲低Cct6a影响mESC的自我更新与多能性的维持Fig.4 Knock-down of Cct6a impairs self-renewal and pluripotency maintenance of mESCs.A:Knockdown efficiency of Cct6a validated by qRT-PCR(**P<0.01 vs shEV group).B:Cell viability of mESCs with Cct6a knockdown (****P<0.0001 vs shEV group).NTC:No template control.C:Morphology of mESCs with Cct6a knockdown;D:Counting number of all clones by colony formation assay(CFA)(**P<0.01 vs shEV group).E:Representative images of CFA.F:Distribution of cell population in G1,S and G2 phases in mESCs with Cct6a knockdown(***P<0.001,**P<0.01,*P<0.05 vs shEV group).
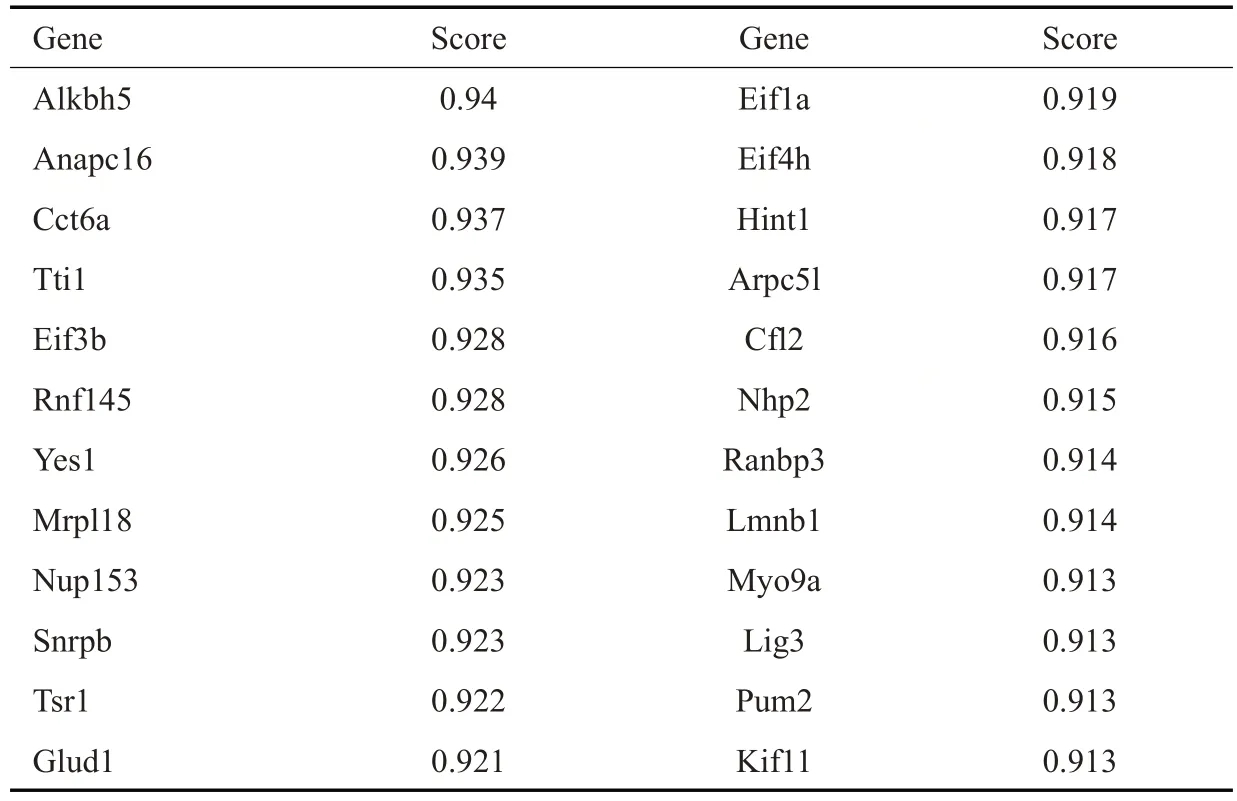
表4 排名最高的部分预测基因Tab.4 Top ranked predicted genes
3 讨论
胚胎干细胞的自我更新与多能性维持的机制与OCT4/SOX2/NANOG作为核心转录因子的转录调控网络紧密相关[38],因此已有的计算生物学方法主要是通过基因的表达,核心转录因子的结合以及与核心转录因子的相互作用来寻找新的自我更新与多能性基因。使用机器学习方法来鉴定胚胎干细胞自我更新与多能性相关基因的研究较少,已在引言部分进行了介绍。研究整合了mRNA微阵列信息和转录因子的ChIP-seq数据,但使用的阳性集仅包含46个基因,这一基因集需要补充最新的结果;虽然其交叉验证最高可获得0.95的AUC[6],但这一研究中的独立测试结果并不理想,阳性测试集中仅有30%~40%的基因被鉴定出。有研究则仅使用了mRNA微阵列信息来寻找mESCs的生物标志物[7],其本质是寻找一群mESCs和其它类型细胞间表达差异最大且能够用来区分细胞类型的基因,属于机器学习中的分类问题而不是进行数据的回归及建立预测模型。Mandal 的模型使用蛋白质相关的特征作为输入,一方面只能鉴定与多能性相关的蛋白质;另一方面由于部分蛋白质相关特征与多能性相关性并不强,模型的表现一般,AUC最高为0.82。
最近的一些研究表明组蛋白修饰、染色质可及性、转录因子结合及染色质三维结构的结构因子结合信息等与小鼠胚胎干细胞的自我更新与多能性基因调控有关,如Nanog 基因通过维持组蛋白第3 亚基4 号赖氨酸的三甲基化(H3K4me3)来诱导自我更新[10];超级增强子可由组蛋白第3 亚基27 号赖氨酸的乙酰化(H3K27ac)来鉴定[11],多能性核心调控因子OCT4,SOX2和NANOG结合到超级增强子位点上激活特定基因的表达[12];ATAC-seq数据可揭示具有增强子和启动子等调控元件的开放性染色质区域[13],在诱导性多能干细胞的重编程过程中,高度开放性的染色质区域与多能性相关,并富集多能性核心调控因子OCT4,SOX2和KLF4(OSK)的结合基序[14];许多介于多能性基因启动子和增强子之间的染色质互作是由结构蛋白CCCTC结合因子(CTCF)[15,16]和黏连蛋白所锚定的[39-41]。阴阳蛋白(YY1)通过与OCT4互作调控多能性基因[17]及增强子与启动子之间的染色质环[9]。上述提到的染色质特征揭示了自我更新与多能性基因与染色质开放性、转录因子和结构因子的染色质结合具有较高相关性,因此多组学整合分析这些染色质特征和基因表达数据,可能是鉴定出潜在的自我更新与多能性基因的一个更好的策略。本研究首次整合了上述多组学数据,使用机器学习方法构建模型预测自我更新与多能性基因。比较多种机器学习方法,最终选择表现最好的随机森林算法,整合多组学数据构建的模型效果比仅使用单一种类数据构建的模型效果更好。最终构建的模型在各项评估指标上水平都较高,在独立测试中取得了较好的效果,曲面下面积达到了0.880±0.028。我们选择了预测出的基因Cct6a进行功能验证,实验结果表明其与ESC的自我更新能力、增殖能力及细胞存活有关。已有的研究表明在癌细胞中CCT6A与SMAD2蛋白有直接结合[42],而在ESC中SMAD2对于primed多能性状态的维持十分重要[43],因此我们猜测CCT6A可能通过SMAD2调控ESC自我更新与多能性,但其调控自我更新、多能性及细胞存活的具体机制仍需进一步研究。
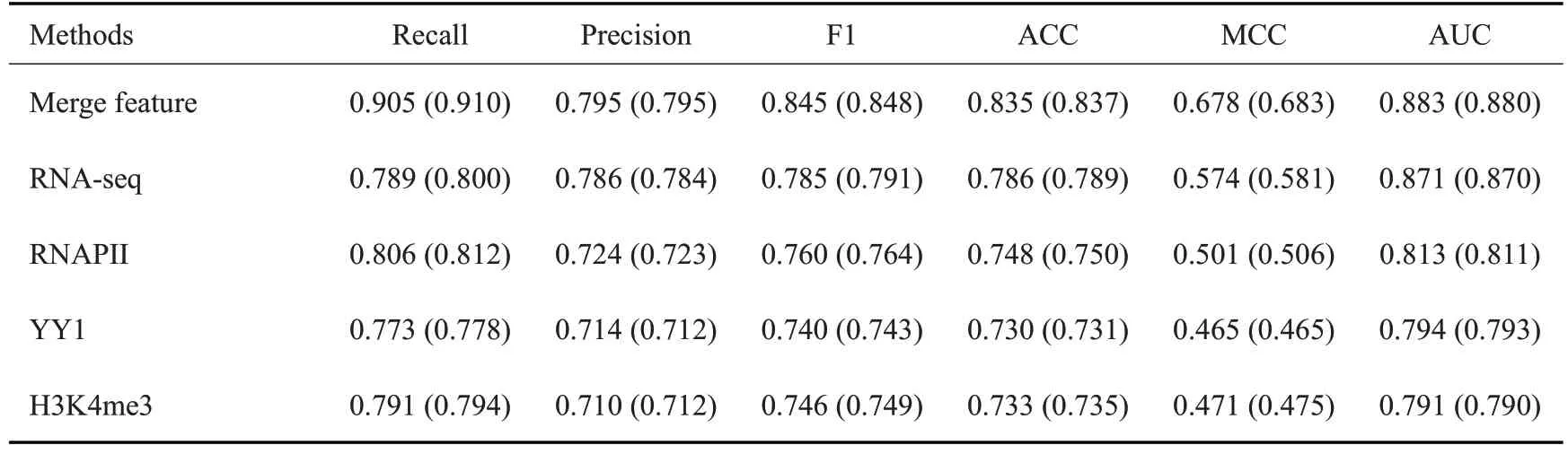
表3 不同特征的交叉验证及独立测试(括号内)的评估指标Tab.3 Performance measures of cross validations and independent tests(in brackets)among different features
综上所述,整合多组学数据可以用于进行胚胎干细胞自我更新与多能性基因的预测,基于随机森林的预测模型具有很好的预测效果。随着单细胞水平高通量测序数据的出现,课题组今后将继续补充可用于进行预测的数据尤其是单细胞数据;另外在模型中补充基因调控网络的信息可以衡量某个基因对于其他基因及整个调控网络的影响进而评估该基因在调控自我更新及多能性方面的重要性。我们将在后续的研究中尝试加入这些信息来改进模型,并进行模型的优化和更大范围的实验验证。
