HPLC法对注射用右兰索拉唑中有关物质的测定
2021-09-08张余霞谭定渊史朝晖
张余霞,谭定渊,史朝晖
(海南碧凯药业有限公司,海口 570311)
随着社会经济的发展,人们生活水平提高以及生活方式的改变,胃肠道疾病的发病率越来越高。2015年在我国城市医院住院患者前10位疾病构成中,消化系统疾病位列第2(占10.14%),而在县级医院中比例达11.10%[1]。消化性溃疡发病原因与胃黏膜屏障受损、黏膜防御/修复机制失衡等因素有关。消化性溃疡在侵蚀胃黏膜血管后,会造成消化道出血或穿孔甚至危及生命[2]。上消化道出血的最常见病因是消化性溃疡,消化性溃疡出血占全部上消化道出血病因的47.43%[3]。
右兰索拉唑为兰索拉唑的右旋单一光学异构体,属于质子泵抑制剂(proton pump inhibitor,PPI),具有生物活性,为抑制胃酸分泌的新型药物。其结构为苯并咪唑类衍生物,作用于二硫键与质子泵(H+-K+-ATP)酶,阻断H+的转运,从而减少胃酸分泌。主要用于非糜烂性胃食管反流病(non-erosive reflux disease,NERD)的“烧心”症状的控制、各种程度的糜烂性食管炎(erosion esophagitis,EE)和糜烂性食管炎愈合期的维持治疗。质子泵抑制剂是目前抑制胃酸分泌强效药物[4]。
2009年,美国FDA批准日本武田制药公司研发的右兰索拉唑缓释胶囊上市[5](商品名为Dexilant®)。2016年,美国FDA批准武田制药美国有限公司的右兰索拉唑缓释口服崩解片上市[6](商品名为Dexilant Solutab)。
右兰索拉唑制剂的质量控制相关方法的报道较少。曾垂宇等[7]建立高效液相色谱法(HPLC)测定右兰索拉唑缓释胶囊的含量测定及其有关物质。对含量和有关物质的分析方法进行方法学验证,结果提示右兰索拉唑与有关物质分离度良好,且空白辅料不干扰检测。郭俊平[8]采用反相高效液相色谱法对右旋兰索拉唑控释胶囊中左旋和右旋异构体进行拆分并测定其中左旋异构体含量,验证结果显示该方法可用于右旋兰索拉唑控释胶囊中左旋异构体的含量测定。张莉华等[9]采用正相高效液相色谱法测定注射用右旋兰索拉唑中左旋异构体的含量,结果提示其方法学验证可行。
本研究在注射用右兰索拉唑处方工艺的基础上,参考《欧洲药典》兰索拉唑有关物质的测定方法[10],建立了注射用右兰索拉唑的高效液相色谱法。
1 材料
1.1 药品与试剂
注射用右兰索拉唑(海南碧凯药业有限公司,批号20160601,规格15 mg,纯度99.8%);右兰索拉唑对照品(加拿大TRC公司,批号2-DXX-95-1,规格100 mg,纯度99.6%);杂质A~E对照品(LGC英国政府化学家实验室,批号:30193、33121、11914、37957、35254,规格100 mg,纯度98.7%、98.4%、95.8%、99.9%、99.9%);三乙胺(罗恩试剂,色谱纯,规格500 ml,纯度99.0%);乙腈(ThermoFisher公式,色谱纯,规格4 L,纯度99.9%);磷酸(广州化学试剂厂,色谱纯,规格500 ml,纯度85.0%)。
1.2 仪器
LC-20AT液相色谱仪(日本岛津仪器设备有限公司,PDA检测器);XS205DU电子分析天平(瑞士梅特勒-托利多公司)。
2 方法与结果
2.1 色谱条件
色谱柱:Agilent Extend-C18柱(250 mm×4.6 mm,5 μm);流动相:三乙胺-水(用磷酸调节至pH 6.2)-乙腈(1∶60∶40);检测波长:285 nm;流速:1.2 ml/min;进样量:10 μl。
2.2 溶液制备
供试品溶液:取注射用右兰索拉唑适量,精密称定,用稀释剂[三乙胺-水(用磷酸调节至pH 10.5)-乙腈(1∶60∶40)]溶解并稀释制成每1 ml中含右兰索拉唑1 mg的溶液,作为供试品溶液。
自身对照品溶液:精密量取供试品溶液适量,用稀释剂定量稀释制成每1 ml中含右兰索拉唑2 μg的溶液,作为对照品溶液。
系统适用性溶液:取右兰索拉唑、杂质B对照品适量,精密称定,加稀释剂超声溶解并稀释制成每1 ml中含右兰索拉唑1 mg及杂质B 40 μg的溶液,作为系统适用性溶液。
2.3 系统适用性试验
取系统适用性溶液10 μl注入液相色谱仪,记录色谱图至主成份峰保留时间的3倍。右兰索拉唑峰与杂质B峰之间的分离度应>3.0。见图1。

1:右兰索拉唑;2:杂质B图1 系统适用性色谱图
2.4 专属性试验
2.4.1空白辅料干扰试验
精密称取空白辅料适量,置于10 ml量瓶中,加稀释剂溶解并稀释至刻度,摇匀,精密量取10 μl注入液相色谱仪。空白辅料溶液在保留时间3 min后无色谱峰出现,不干扰本品的有关物质测定。见图2。

图2 空白辅料色谱图
2.4.2供试品溶液
取注射用右兰索拉唑适量,精密称定,用稀释剂溶解并稀释制成每1 ml中含右兰索拉唑1 mg的溶液,作为供试品溶液。在“2.1”的色谱条件下进样,记录色谱图。见图3。

图3 注射用右兰索拉唑供试品图谱
2.4.3已知杂质对照品混合溶液
精密称取右兰索拉唑、起始物料(氯化物,见图4“6”注释)、杂质A~E对照品适量,分别加少量稀释剂超声溶解并用稀释剂稀释至刻度,摇匀,作为储备液;精密移取各储备液1 ml置同一10 ml量瓶中,加稀释剂稀释至刻度,摇匀,作为混合对照品溶液,取10 μl注入液相色谱仪,记录色谱图。结果显示,各杂质峰、起始物料峰、右兰索拉唑峰之间的分离度均>1.5,达到系统适用性要求。见图4。

1:杂质D;2:杂质E;3:杂质A;4:右兰索拉唑;5:杂质B;6:氯化物[2-氯甲基-3-甲基-4-(2,2,2-三氟乙氧基)-2-吡啶盐酸盐];7:杂质C图4 混合对照品溶液色谱图
2.5 检测限和定量限
分别取右兰索拉唑与杂质A~E对照品适量,精密称定,用稀释剂超声溶解并用稀释剂稀释制成一定浓度的溶液作为储备液。采用逐步稀释法进行试验,以信噪比不小于10为定量限,信噪比不小于3为检测限。测定结果见表1。
2.6 线性和范围
精密称取杂质A~E、右兰索拉唑对照品各约10 mg,分别置于50 ml量瓶中,精密称取杂质B约10 mg,置于20 ml量瓶中,加稀释剂适量溶解并稀释至刻度,摇匀,分别作为杂质A~E、右兰索拉唑对照品的储备液;分别精密量取杂质A、C、D、E、右兰索拉唑对照品储备液1 ml,杂质B对照品储备液2 ml,置同一10 ml量瓶中,加稀释剂稀释至刻度,摇匀,作为混合对照品储备液。分别精密量取混合对照品储备液适量,用稀释剂稀释成溶液杂质A~E 20、40、20、20和20 μg/ml,分别精密量取上述溶液各 10 μl 注入液相色谱仪,记录色谱图,测量峰面积。以峰面积y为纵坐标,进样浓度x为横坐标,做线性回归曲线(线性回归时以定量限结果为线性起始点)。测定结果见表2~3。

表2 标准曲线 n=6

表3 杂质的校正因子
校正因子=Slope主药/Slope杂质
结果可知,杂质B~D的校正因子在0.9~1.1范围内,可以用主成份自身对照法计算含量;杂质A和E的校正因子分别为0.60和0.34,可用加校正因子的主成份自身对照法计算杂质A和E的含量。
采用主成份自身稀释对照法计算有关物质含量,计算公式:


总杂质计算公式:总杂质(%)=杂质A(%)+杂质B(%)+杂质C(%)+杂质D(%)+杂质E(%)+其他杂质(%)
其中,AX:供试品中杂质的峰面积;AR:对照品溶液中主峰的峰面积。
2.7 准确度试验
精密称定杂质A~E对照品各适量,用稀释剂分别溶解并稀释,制成杂质A、C、D、E浓度为200 μg/ml的溶液,杂质B为400 μg/ml的溶液,摇匀,作为各杂质对照品储备液;分别精密量取各杂质对照品储备液0.8、1和1.2 ml,置于10 ml量瓶中,同时精密量取供试品适量(相当于右兰索拉唑10 mg),分别置于上述10 ml量瓶中,加稀释剂稀释至刻度,作为80%、100%和120%回收率样品溶液;每个浓度平行制备3份。精密量取杂质对照品储备液1 ml,置于10 ml量瓶中,加稀释剂稀释至刻度,摇匀,作为对照品溶液。
分别取上述杂质对照品溶液、供试品溶液及回收率样品溶液各10 μl,注入液相色谱仪,记录色谱图,采用外标法计算杂质回收率。回收率测定结果见表4~8。
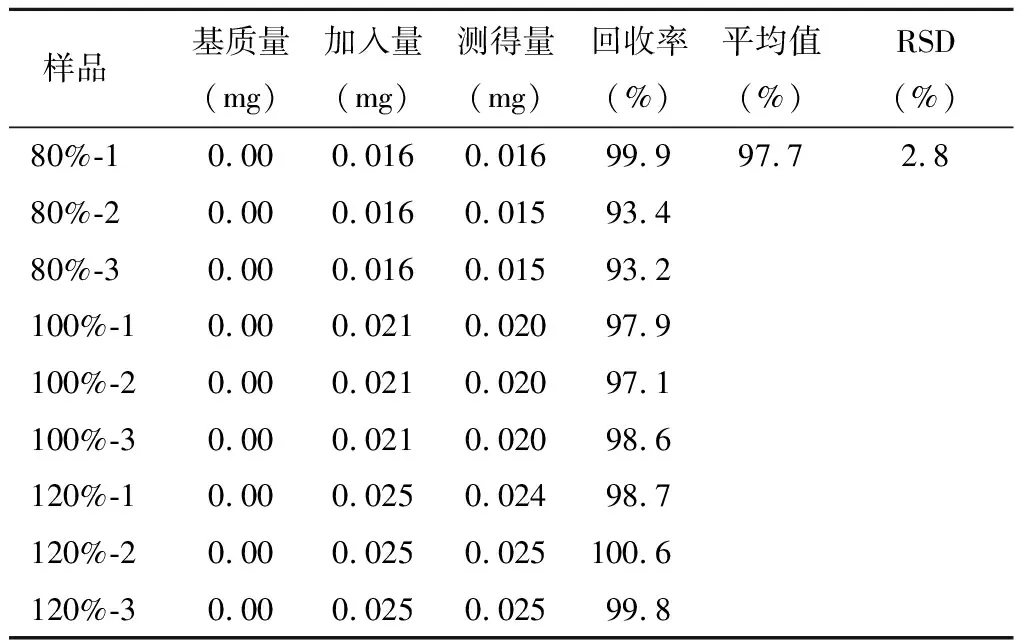
表4 杂质A回收率测定结果 n=9

表5 杂质B回收率测定结果 n=9

表6 杂质C回收率测定结果 n=9

表8 杂质E回收率测定结果 n=9
结果显示,各杂质的回收率均符合要求,准确度良好。
2.8 精密度试验
2.8.1重复性
取注射用右兰索拉唑10瓶,取出内容物,混匀,精密称定粉末(相当于右兰索拉唑10 mg),加适量稀释剂溶解,并稀释制成每1 ml含右兰索拉唑约 1 mg 的溶液,摇匀,滤过,取续滤液作为供试品溶液。精密量取适量,用稀释剂稀释成每1 ml中含右兰索拉唑 2 μg 的溶液作为对照品溶液,平行配制6份。取上述方法制备的对照品溶液和供试品溶液各10 μl,进样测定,记录色谱图,计算杂质含量,测定结果见表9。
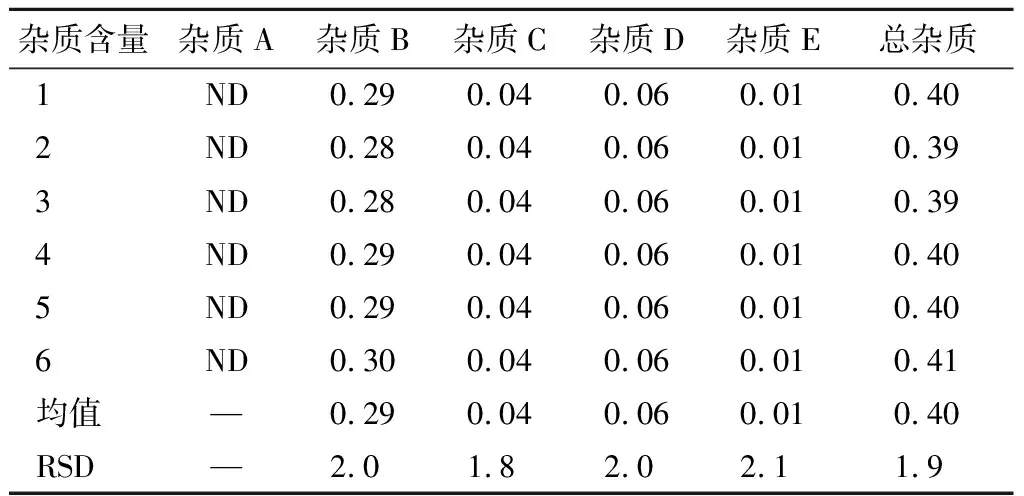
表9 重复性测定结果 %
ND:未检出。下表同。
结果显示,本方法的重复性良好。
2.8.2中间精密度
由不同分析人员在不同日期、不同仪器上测定同一批注射用右兰索拉唑样品杂质的含量,考察中间精密度。见表10。

表10 中间精密度测定结果 %
中间精密度:仪器二、分析人员B
结果显示,12个样品的杂质A均未检出;杂质B的平均值为0.28%,RSD为2.3%;杂质C的平均值为0.04%,RSD为3.0%;杂质D的平均值为0.06%,RSD为2.5%;杂质E的平均值为0.01%,RSD为3.0%;总杂质的平均值为0.39%,RSD为2.3%。表明本方法中间精密度良好。
2.9 溶液稳定性试验
取重复性第1份样品,作为供试品溶液;精密量取适量,用稀释剂稀释制成每1 ml中含右兰索拉唑2 μg的溶液作为对照品溶液。在室温条件下放置0、2、4、6和8 h,分别精密吸取供试品溶液与对照品溶液各10 μl,注入高效液相色谱仪,计算杂质含量。见表11。
结果表明,供试品溶液在室温条件下放置8 h,稳定性良好。
2.10 耐用性试验
取注射用右兰索拉唑10瓶,取出内容物,混匀,精密称定粉末(相当于右兰索拉唑10 mg),加适量稀释液,超声溶解后,用稀释溶液稀释成每1 ml含右兰索拉唑约1 mg的溶液,摇匀,过滤,取续滤液作为供试品溶液;精密量取适量供试品溶液,用稀释溶液稀释成每1 ml中含右兰索拉唑2 μg的溶液作为对照品溶液。分别考察乙腈比例变化±2%,流速变化±0.1 ml/min,流动相pH±0.2,柱温变化±5 ℃及不同品牌色谱柱条件下,杂质B与主峰的分离度。见表12。

表12 耐用性试验结果
结果显示,各条件下主成份峰与杂质B峰分离度均>3.0。表明该方法的耐用性良好。
2.11 样品测定
取3批注射用右兰索拉唑(批号20160601、20160602、20160701),按“2.2”项下方法制备供试品溶液,按“2.1”项下色谱条件进样分析。见表13。
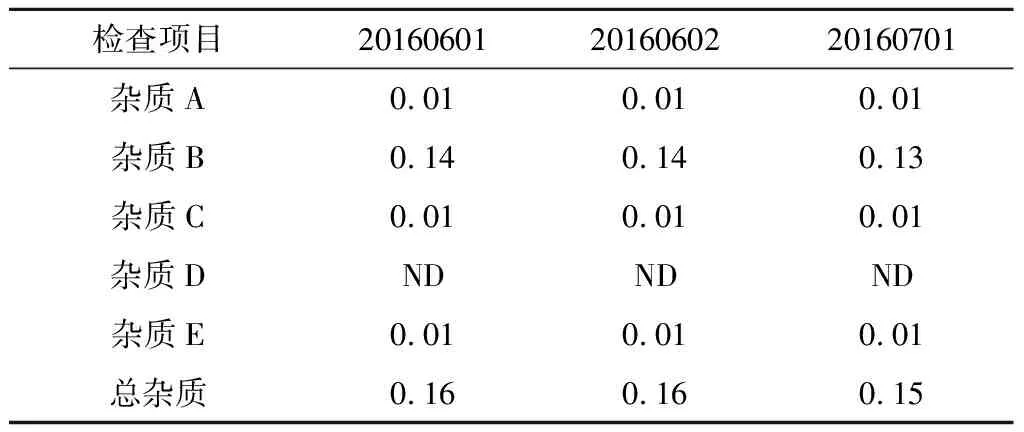
表13 有关物质测定结果 %
经方法学验证,有关物质的检查方法可行。
3 讨论
本研究建立了HPLC法测定注射用右兰索拉唑有关物质的方法,确定色谱条件并进行了方法学考察。经方法学验证,表明方法专属性强、准确度高、重复性好,适用于注射用右兰索拉唑中有关物质的检测。
有关物质分析方法考虑的杂质间及与主峰间分离度需达到有关要求,通过系统的方法开发,考虑到杂质B与右兰索拉唑主峰的相对保留时间较为接近,故系统适用性溶液以右旋兰索拉唑与杂质B的分离度应>3.0作为系统通过的判定条件。
取适量注射用右兰索拉唑用稀释液溶解并稀释成含右兰索拉唑10 μg/ml的溶液,在紫外200~400 nm的波长下进行扫描,其在210及285 nm附近均有最大吸收,但在285 nm波长处时,供试品检出杂质个数及量均高于210 nm,且制剂中其他辅料峰位于低紫外210 nm波长处干扰明显高于285 nm波长,所以最终285 nm作为本品有关物质的控制检测波长。
流动相参考《中国药典》[11]兰索拉唑标准,选择水及乙腈作为主要溶剂,通过加入三乙胺及磷酸调节pH来控制供试品中各物质存在的离子形式,避免峰型拖尾,确保积分准确。
