阿司匹林中对羟基苯甲酸杂质测定不确定度评定方法的建立
2021-09-08庞发根李玉兰王铁杰
庞 赛,庞发根,江 坤,李玉兰,王铁杰
(深圳市药品检验研究院,深圳市药品质量标准研究重点实验室,深圳 518057)
欧洲药品质量管理局(EDQM)是目前全球最权威的实验室能力验证机构之一,对实验室独立完成检测的能力有较高的要求。测量不确定度是表征赋予被测量值分散性的非负参数,一个完整的定量分析结果应包含测量值和测量不确定度2个指标[1-2]。近年来,不确定度在药物分析领域中的应用越来越广泛[3-8],如今在EDQM的能力验证中,组织方亦要求参与者报告测定结果的不确定度值,让能力验证的参与者能够将自己的不确定度估计值与其他实验室的估计值进行比较。因此,测量不确定度的评定逐渐引起了广大药物分析人员的重视。正确表达和评定测量方法的测量不确定度将逐步成为国际通用的要求。
阿司匹林为非甾体抗炎药,主要的治疗功效是解热、镇痛、抗风湿、抑制血小板聚集等作用。该品种在各国药典中均有收载,对羟基苯甲酸为《欧洲药典》[9]阿司匹林标准中的特定杂质A。本研究为本实验室在参加EDQM液相色谱法能力验证中以《欧洲药典》阿司匹林标准项下收载的方法对杂质对羟基苯甲酸的含量进行测定,参考相关资料并结合EDQM测量不确定度评定的核心文件[10],对《欧洲药典》阿司匹林中的对羟基苯甲酸测定的不确定度进行了分析,以期为有效控制对羟基苯甲酸测定方法的准确性,以及对今后各实验室参加EDQM能力验证时评定不确定度提供参考。
1 药品与试剂
对羟基苯甲酸对照品(由EDQM能力验证组织方提供,含量为100.0%);乙腈为色谱级;磷酸为分析级;超纯水为实验室自制。
安捷伦1260高效液相色谱仪(美国安捷伦公司);XS205DU电子分析天平(瑞士梅特勒-托利多仪器公司);10 ml量瓶(A级)、25 ml量瓶(A级)、100 ml量瓶(A级)、1 ml单标移液管(A级)。
2 实验方法
测定方法:依据《欧洲药典》10.0版阿司匹林有关物质项下杂质A的测定方法及EDQM能力验证文件ProficiencyTestingSchemeonLiquidChromatography(PTS208)[11]。
色谱条件:采用以十八烷基硅烷键合硅胶为填充剂的色谱柱,以磷酸-乙腈-水(2∶400∶600)为流动相,检测器为紫外检测器,检测波长237 nm,进样量为10 μl,流速为1.0 ml/min。
对照品溶液配制:取对羟基苯甲酸对照品约 50 mg,精密称定,置于25 ml量瓶中,用流动相稀释至刻度,摇匀,精密量取1.00 ml该溶液至100 ml量瓶中,用流动相稀释至刻度,摇匀。
供试品溶液配制:取阿司匹林供试品约0.1 g,精密称定,置于10 ml量瓶中,用流动相稀释至刻度,摇匀。
样品测定:取对照品溶液、供试品溶液各10 μl,分别注入高效液相色谱仪并测定峰面积,按外标法计算对羟基苯甲酸的含量。
3 测量模型的建立
按公式计算对羟基苯甲酸含量:
则:
其中,X为供试品中对羟基苯甲酸含量(%);CT为供试品浓度(mg/ml);CS为对照品浓度(mg/ml);WT为供试品的取样量(g);WS为对照品的取样量(g);AT为供试品溶液色谱图中对羟基苯甲酸的峰面积;AS为对照品溶液的主峰面积;VT为供试品的稀释体积(ml);VS为对照品的稀释体积(ml);ωS为对照品的含量(%)。
实际上X的测得值是3份平行试样的测定数据给出的,重复性标准不确定度不可忽略。应加入重复性修正量R,其值视为零,不确定度非零。加入后公式变为:
X′=X+R
其中,X′为考虑重复性修正量R后的实际含量(%);X为供试品含量(%);R为重复性修正量(%)。
4 不确定度来源分析
不确定度来源用因果图表示,见图1~图3。

图1 X的不确定度来源因果图

图2 VS的不确定度来源因果图

图3 加入重复性后的不确定度来源因果图
5 评定对羟基苯甲酸含量的标准不确定度[u(X)]
5.1 评定对照品称量的相对标准不确定度[urel(WS)]
5.1.1评定u(WS)
u(WS)由天平示值误差的不确定度u1(WS)和天平称量重复性的不确定度u2(WS)合成,均属于B类测量不确定度。
故:
①u1(WS):对照品称量约为50 mg,查天平(XS205DU)检定证书,所用电子天平(d=0.01 mg)的最大允差为±0.05 mg,认为矩形分布:
②u2(WS):称量对照品所用天平的检定证书给出的重复性误差为0.01 mg,认为矩形分布:
则单次称量的标准不确定度:
实际实验过程使用减重法,故不确定度为:
5.1.2评定urel(WS)
对照品称样量分别为0.050 10和0.049 85 g,则相对标准不确定度:
=1.177×10-3(mg)
5.2 评定供试品称量的相对标准不确定度[urel(WT)]
5.2.1评定u(WT)
u(WT)由天平示值误差的不确定度u1(WT)和天平称量重复性的不确定度u2(WT)合成,均属于B类测量不确定度。
故:
①u1(WT):供试品称量约为0.1 g,查天平(XS205DU)检定证书,所用电子天平(d=0.01 mg)的最大允差为±0.05 mg,认为矩形分布:
②u2(WT):称量对照品所用天平的检定证书给出的重复性误差为0.01 mg,认为矩形分布:
则单次称量的标准不确定度:
实际实验过程使用减重法,故不确定度为:
5.2.2评定urel(WT)
对照品称样量分别为0.099 51、0.100 20和0.099 70 g,则相对标准不确定度:
urel(WT)=0.722×10-3(mg)
5.3 评定对照品峰面积的相对标准不确定度[urel(AS)]
对照品峰面积的相对标准不确定度主要来源为进样重复性,属A类不确定度,取2份对照品溶液分别连续进样3次,对照品峰面积测定结果见表1,用极差法计算对照品峰面积的标准不确定度。

表1 对照品峰面积测定结果
=0.494×10-3
=0.383×10-3
则对照品峰面积的相对标准不确定度:
5.4 评定供试品峰面积的相对标准不确定度[urel(AT)]
2份供试品溶液分别连续进样2次,峰面积测定结果见表2,用极差法计算供试品峰面积的标准不确定度。

表2 供试品峰面积测定结果
=2.546×10-3
=2.157×10-3
=1.453×10-3
则供试品峰面积的相对标准不确定度:
=3.639×10-3
5.5 评定对照品稀释体积的相对标准不确定度[urel(VS)]
对照品溶液稀释过程的不确定度由25 ml量瓶引入的不确定度u(VS1)和100 ml量瓶引入的不确定度u(VS3)、1 ml单标移液管引入的不确定度u(VS2)合成,分别计算每一步稀释过程引入的不确定度。
采用相对标准不确定度计算:
5.5.1评定urel(VS1)
u(VS1)由25 ml量瓶最大允差引起的标准不确定度u1(VS1)和温度效应引入的标准不确定度u2(VS1)合成。
①u1(VS1):在初始溶解对照品时,用到25 ml量瓶,查量瓶检定规程JJF196—2006《常用玻璃量器》,使用的25 ml量瓶最大允许误差绝对值为0.03 ml,历年检定结果显示误差均较小,视为三角分布:
②u2(VS1):实验在控温环境下进行,室温控制在(20±5) ℃,水的体积膨胀系数为0.000 21/℃,按操作描述得初步定容体积为25 ml。因此控温范围产生的体积增量半宽分别为25×0.000 21×5=0.026 25(ml)。因为计算结果时不做温度体积修正,此处将体积变化作为不确定度处理,温度导致的体积变化为矩形分布,则其标准不确定度为:
合成得到:
则:

5.5.2评定urel(VS2)
u(VS2)由1 ml单标移液管最大允差引起的标准不确定度u1(VS2)和温度效应引入的标准不确定度u2(VS2)合成。
①u1(VS2):对照品两步稀释时,用到1 ml单标移液管,查检定规程JJF196—2006《常用玻璃量器》,使用的A级1 ml单标移液管最大允许误差绝对值为0.007 ml,历年检定结果显示误差均较小,视为三角分布:
②u2(VS2):实验在控温环境下进行,室温控制在(20±5) ℃,水的体积膨胀系数为0.000 21/℃,按操作描述吸液体积为1 ml。因此,控温范围产生的体积增量半宽分别为1×0.000 21×5=0.001 05(ml)。因为计算结果时不做温度体积修正,此处将体积变化作为不确定度处理,温度导致的体积变化为矩形分布,则其标准不确定度为:
合成得到:
=2.914×10-3(ml)
则:
=2.914×10-3(ml)
5.5.3评定urel(VS3)
u(VS3)由100 ml量瓶最大允差引起的标准不确定度u1(VS3)和温度效应引入的标准不确定度u2(VS3)合成。
①u1(VS3):对照品第2步稀释时,用到100 ml量瓶,查量瓶检定规程JJF196—2006《常用玻璃量器》,使用的100 ml量瓶最大允许误差绝对值为0.10 ml,历年检定结果显示误差均较小,视为三角分布:
②u2(VS3):实验在控温环境下进行,室温控制在(20±5)℃,水的体积膨胀系数为0.000 21/℃,按操作描述得初步定容体积为100 ml。因此,控温范围产生的体积增量半宽分别为100×0.000 21×5=0.1050(ml)。因为计算结果时不做温度体积修正,此处将体积变化作为不确定度处理,温度导致的体积变化为矩形分布,则其标准不确定度为:
合成得到:
则:

对照品溶液稀释的相对标准不确定度:
=3.104×10-3(ml)
5.6 评定供试品稀释体积的相对标准不确定度[urel(VT)]
u(VT)由10 ml量瓶最大允差引起的标准不确定度u1(VT)和温度效应引入的标准不确定度u2(VT)合成。故:
①u1(VT):溶解供试品时用到10 ml量瓶,查量瓶检定证书及根据检定规程JJF196—2006《常用玻璃量器》,使用的10 ml量瓶最大允许误差绝对值为0.020 ml,视为三角分布:
②u2(VT):实验在控温环境下进行,室温控制在(20±5) ℃,与20 ℃的温差导致的溶液体积膨胀大于量瓶的体积膨胀,可只考虑溶液的体积膨胀。水的体积膨胀系数为0.000 21/℃,按操作描述定容体积为10 ml。因此控温范围产生的体积增量半宽分别为10×0.000 21×5=0.010 50(ml)。因为计算结果时不做温度体积修正,此处将体积变化作为不确定度处理,假设温度导致的体积变化为矩形分布,则其标准不确定度为:
合成得到
则:
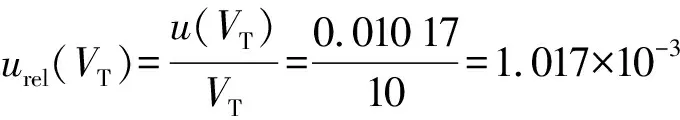
5.7 评定对照品含量的相对标准不确定度[urel(ωS)]
对羟基苯甲酸对照品(EDQM,含量:100.0%)无相关的不确定度信息,且实验室目前无同类具有不确定度信息的对照品替代,故忽略urel(ωS)。
5.8 对羟基苯甲酸含量的标准不确定度u(X)
对羟基苯甲酸含量的相对标准不确定度:

实验测得对羟基苯甲酸含量为3份平行样的平均值,X=0.250%
故:
u(X)=urel(X)×X=5.120×10-3×0.250%=0.001 28%
6 评定重复性的标准不确定度[u(R)]
重复测定3份平行样,测定结果见表3。

表3 重复性结果 %
用贝塞尔公式计算标准差:
规程中要求测定3份平行样,则:
7 计算合成标准不确定度
合成标准不确定度,见公式:
8 报告测量结果
阿司匹林供试品中对羟基苯甲酸含量结果为:(0.250±0.002)%,第2项为标准不确定度(非扩展不确定度)。不确定度一览表见表4。

表4 不确定度评定一览表
不确定度A类评定:用对观测列进行统计分析的方法来评定标准不确定度;不确定度B类评定:用不同于对观测列进行统计分析的方法来评定标准不确定度
9 讨论
测量不确定度评定应尽可能分析测量不确定度的所有可能来源,本文通过参考EDQM测量不确定度评定核心文件,对《欧洲药典》[9]收载的阿司匹林标准中对羟基苯甲酸测定过程中的不确定度各分量来源进行分析,建立了测定阿司匹林中对羟基苯甲酸的不确定度评定方法,可为同分析方法的不确定度评定提供参考。
由表4可知,本研究不确定度分量中,对结果影响最大的分量为测量重复性。因此,在整个实验过程中应注重分析人员平行操作的重要性,在双份样品或多份样品测定时,应将测量结果的相对标准偏差控制在要求的范围内,以保证数据的准确性和可靠性。提示可通过以下途径降低不确定度,提高检验结果的准确性和可靠性:① 定期对液相色谱仪进行性能确认,加强日常维护保养,确保仪器始终处于稳定的工作状态并具有良好的进样精密度。② 在分析过程中,溶液稀释过程应使用经校准合格的容量仪器,合理控制实验环境温度。③ 称量应选用经性能确认合格的天平,且天平精度符合使用要求。④ 实验人员在整个测定过程中应注意标准操作,避免引起不必要的误差,使测定结果更加准确可靠。
