助催化剂强化电芬顿技术去除水中难降解有机物的研究进展
2021-09-07郑豪程松沈晨于伟华刘福强
郑豪,程松,沈晨,于伟华,刘福强,
(1. 南京大学 环境学院;污染控制与资源化研究国家重点实验室,南京 210023;2. 南京环保产业创新中心有限公司,南京 211106)
化工、医药、农药等行业生产过程中产生大量含多环芳烃、卤代烃、杂环类化合物、农药、抗生素等难降解有机物的工业废水,通常具有高毒性、高盐分等特征,已成为工业水处理领域的难点[1-2]。
对于工业废水中难降解有机物,通常采用氧化法、物化法等技术处理。近年来,高级氧化技术因作用效率高、适用范围广等优势而获得快速发展,其中以羟基自由基(HO·)为主导的芬顿氧化技术受到持续关注,但因H2O2利用率低、Fe2+用量大以及大量铁泥后续处置难等问题,限制了其进一步应用推广[3]。
电芬顿技术将电化学与芬顿技术相结合,主要优势包括:可原位生成H2O2,降低H2O2在运输、存储等过程的安全风险,缓解了一次性投加H2O2导致的自分解问题;阴极提供电子,将Fe3+有效还原为Fe2+,大幅减少铁泥产量;实现阳极氧化、电吸附等协同作用,显著提高有机物去除效率[4-6]。
笔者基于电芬顿原理、局限性以及强化方法,着重分析助催化剂强化电芬顿技术的原理、特性并展望了改良方向。
1 电芬顿及其强化方法
1.1 电芬顿原理
电芬顿技术以Fe2+和电化学产生的H2O2作为芬顿试剂的来源[7],通过Fe2+与H2O2发生芬顿反应生成HO·,从而实现污染物的高效降解,如式(1)~式(5)所示。

(1)

(2)
(3)
Intermediate products
(4)
CO2+H2O+Inorganic ions
(5)
电芬顿可以分为均相电芬顿和非均相电芬顿两大类。其中,均相电芬顿中Fe2+与H2O2在溶液中发生均相反应产生活性氧物种,而非均相电芬顿则以铁活性物种或将其负载至载体作为铁源催化剂,在其表面发生非均相反应并产生自由基。
电芬顿去除难降解有机物的效率主要受3方面因素限制:1)pH值适用范围窄,通常需要酸性条件(pH≈3.0);2)阴极H2O2产率和电流效率低,限制了芬顿反应速率;3)Fe3+/Fe2+的循环速率是生成羟基自由基降解污染物的主要限速步骤。
1.2 电芬顿强化方法
为提高电芬顿技术对难降解有机物的去除效率,研究和应用较多的强化方法主要包括3方面:1)优化设计反应器,通过增强传质使污染物与活性氧充分接触[4];2)载体固定催化剂,能够提高催化剂的稳定性、减少金属离子的浸出,同时,催化剂分散在载体上,活性位点增加,可以提高催化活性;3)添加助催化剂,包括直接投加到电解液中或同催化剂一起制备到阴极上,能有效增加Fe2+的含量或提高H2O2的选择性生成,提高催化剂的活性、稳定性等。
近年来大量研究发现,添加少量助催化剂即可有效提升电芬顿对难降解有机物的去除效果[8-11]。研究较多的3种助催化手段及其机理如表1所示。

表1 电芬顿助催化手段及机理Table 1 Co-catalysis methods and mechanisms of EF
2 助催化剂强化电芬顿技术
电芬顿涉及的催化反应主要为阴极催化O2通过两电子氧还原反应(2e-ORR)生成H2O2以及Fe2+催化H2O2生成HO·。为增强催化剂的催化作用,可通过优选优用助催化剂,直接或间接增强电芬顿技术性能。
2.1 改善铁离子稳定性
铁在较高pH值条件下会沉淀形成大量铁泥,导致有效铁含量减少,进而削弱污染物的去除效率。铁离子与配体的配位作用可以保证其在接近中性pH的条件下以离子形态存在,因此,添加配体能拓宽有效pH范围。而且,络合物可通过一系列反应产生更多自由基,从而增强电芬顿去除有机污染物的性能。其反应机理如图1所示。
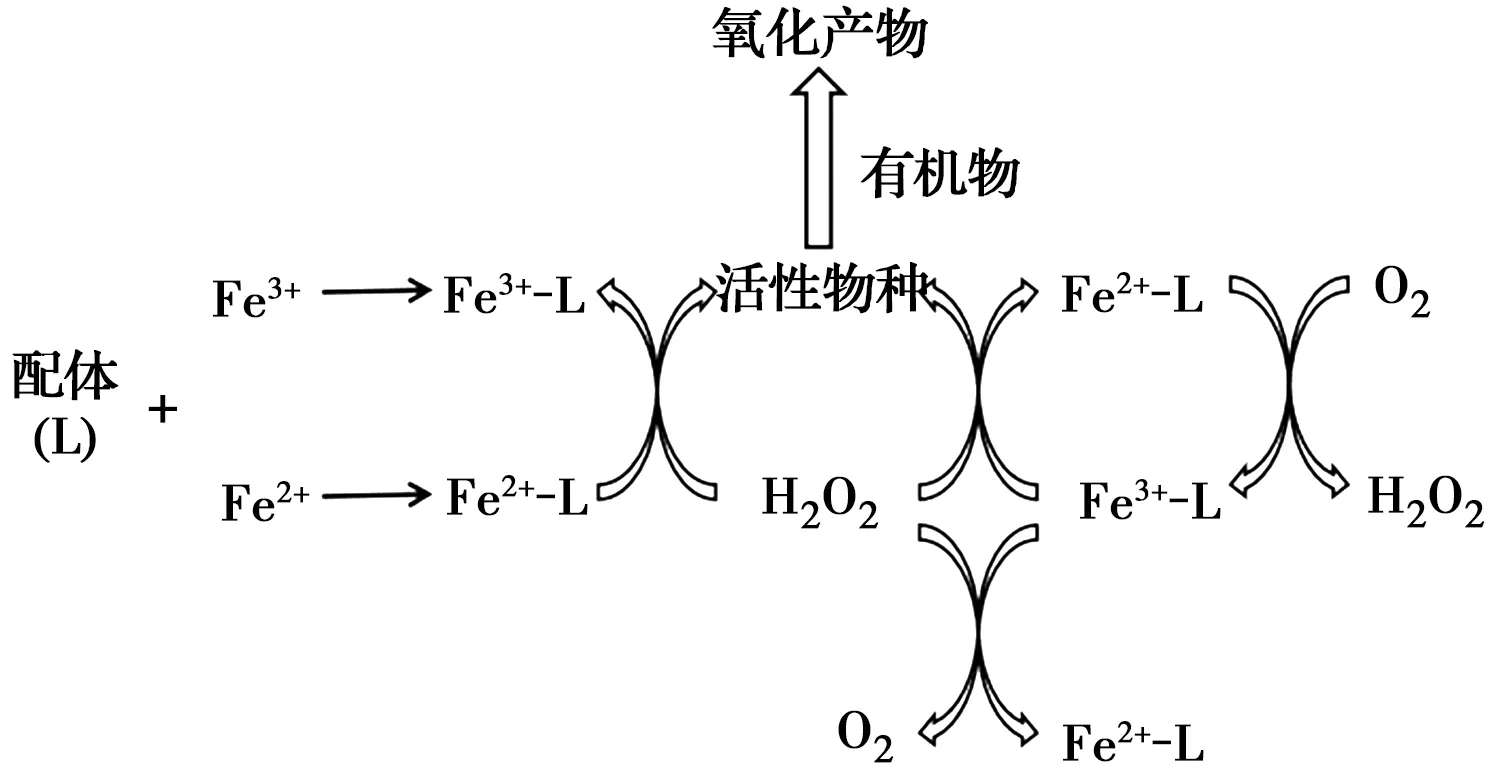
图1 配体强化芬顿反应机理Fig.1 Mechanism diagram of ligand enhanced
乙二胺四乙酸(EDTA)、氮三乙酸(NTA)、乙二胺二琥珀酸(EDDS)、羟胺等有机配体被广泛应用到芬顿体系中,可在pH>3.0的条件下有效去除难降解有机物。Zhang等[12]将NTA加入电芬顿体系,pH=5.0~8.0下均可在20 min内完全去除水中苯酚,速率常数约为0.26 min-1,较不加助催化剂提高了近63%。强络合剂EDDS是一种具有生物降解性的有机配体,与NTA相比,对环境的二次污染更小,Ye等[13]将其引入电芬顿体系,以Fe(Ⅲ)-EDDS为可溶性催化剂,pH=9.0下仅约6%的铁发生沉淀,而未添加EDDS时89%以上的铁发生沉淀。不同的Fe-L(配体)络合物的氧化还原性能存在差异,Liu等[14]结合理论计算发现,[Fe(Ⅲ)-EDTA]-络合物的电子还原速率约是[Fe(Ⅲ)-EDDS]-络合物的两倍,更强的得电子能力有助于加速铁循环,进而促进电芬顿性能。除了拓宽pH值范围外,部分有机配体不仅作为螯合剂,同时作为还原剂,利用配体-金属间的电荷转移,促进Fe3+还原为Fe2+,从而增强电芬顿效率[15-17],Hou等[18]发现在芬顿体系中添加羟胺后HO·的生成速率常数是未添加时的100~10 000倍。然而,有机配体投加会增加有机负荷,进一步增加处理成本。此外,在处理过程中产生的活性物质会氧化分解有机配体,从而降低活性及稳定性[19]。
为此,近年来无机配体颇受重视。Wang等[20]用四聚磷酸钠(Na6TPP)作为电芬顿体系电解质,形成的铁-四聚磷酸盐络合物(Fe-TPP)保证铁以离子形态存在于溶液中,可以在pH值4.0~10.2的范围内有效降解阿特拉津(ATZ)。聚磷酸盐虽然不会消耗HO·,但其使用会导致后续除磷难题。Cui等[21]采用环境友好的无机配体二硅酸钠(SD)增强铁电解系统,Fe2+/Fe3+-SD络合物的生成有效避免了铁离子的水解沉淀,因此,可在pH值为5.0~8.0条件下稳定运行。另一方面,铁离子与SD络合,可以有效降低Fe3+/Fe2+的氧化还原电位,使O2的还原热力学过程更有利[22]。
综上,多种有机和无机配体均可有效拓宽电芬顿体系pH适应范围,并在近中性条件下获得优良去除效果,如表2所示。然而,铁配合物的重复利用以及后续处理问题亟待解决。Jin等[23]发现用载体固定铁络合物可提高重复利用率,通过将铁-二吡啶甲酰胺络合物(Fedpa)固定在SiO2上成功制备Fedpa@SiO2催化剂,重复利用3次后,仍能去除90%以上的2,4-二氯苯酚(2,4-DCP)。
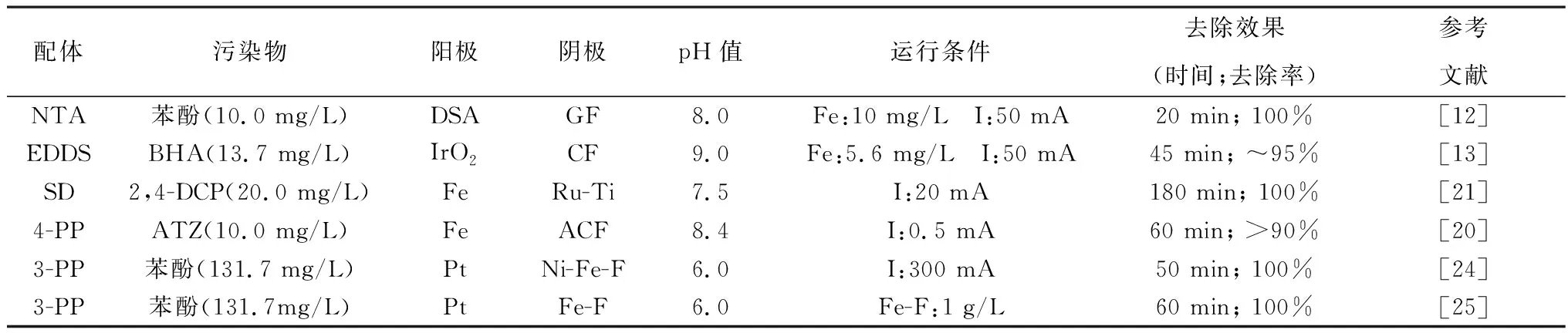
表2 配体强化电芬顿体系去除难降解有机物的效果Table 2 Effect of ligand enhances EF system in removing refractory organics
目前,对于配体强化电芬顿技术的研究仍集中在单一配体的作用机制。实际废水水质复杂,通常同时存在多种有机配体和无机配体,阐明不同配体之间的相互作用规律与机制,可为直接利用水体中存在的配体提供重要参考,进而减少外源投加,降低可能造成的二次污染风险。
2.2 提升Fe3+/Fe2+循环速率
Fe3+还原为Fe2+的速率直接影响了电芬顿中HO·的生成速率以及污染物的降解速率,因此,加速Fe3+/Fe2+循环一直是研究热点。添加能够改善催化剂电子传递性能的助催化剂,可以加速Fe2+的再生。目前研究较多的是双金属协同催化技术。
在Fe-M(M为其他金属)双金属体系中,利用双金属协同效应可以促进Fe2+再生,机理图如图2所示。金属硫化物是研究较多的助催化剂,其表面不饱和的S原子捕获质子形成H2S后,暴露具有还原性的金属活性位点,从而加速Fe3+/Fe2+循环,Li等[26]在电芬顿体系中投加WS2后,罗丹明B(RhB)降解的速率常数提升了63.1%,这得益于W4+快速还原Fe3+。Mo是工业上常用的低毒性催化剂,Tian等[27]用MoS2助催化电芬顿技术去除磺胺甲嘧啶(SMT),污染物去除的表观速率常数从原先的0.26 min-1增加到0.54 min-1。往反应池中直接投加助催化剂存在难以重复利用、增加运行成本的问题,因此,将过渡金属与Fe一起制备成复合材料得到了学者们的关注。
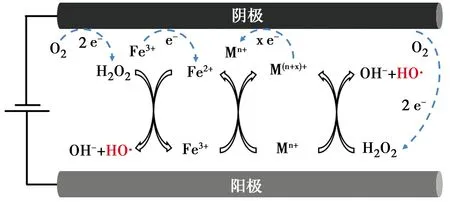
图2 Fe-M双金属催化剂电芬顿体系机理Fig.2 Mechanism diagram of electro-Fenton EF system with
Cu是一种导电性好、廉价的过渡金属,因而应用广泛,多种铁铜双金属催化剂被成功用于电芬顿体系,如表3所示。Luo等[28]通过两步还原法制备了Cu掺杂的Fe@Fe2O3(CFF)纳米粒子,Cu/Fe质量比为50%时,2 h内四环素(TC)去除率较nZVI提升了近12%。Barros等[29]通过共沉淀法合成Fe3-xCuxO4(0≤x≤0.25)NPs,由于铁和铜离子在尖晶石结构八面体表面位置的协同作用,Cu2+/Cu+表面物种的存在对紫红花食用染料的降解有显著促进作用,Fe2.75Cu0.25O4体系的TOC去除率远高于Fe3O4体系,前者约为70%,而后者仅不足39%。由于Cu本身也能催化H2O2生成HO·,因此,铁铜协同作用的相对贡献需要进一步计算。Ren等[30]利用一种新亚铜试剂捕获Cu(Ⅰ),屏蔽Cu(Ⅰ)与铁和H2O2的相互作用,从而定量鉴别各因素对增强芬顿反应催化活性的贡献,其中19%的增强作用被确定为铁铜协同作用。Co是一种具有类芬顿活性的过渡金属,Ganiyu等[31]在碳毡(CF)上生长了分级CoFe-层状双氢氧化物(CoFe-LDH)并制成阴极,可在较宽的pH值范围内(pH值为2.0~7.1)实现酸性橙II (AO7)的有效矿化,在pH值为3.0的条件下2 h内TOC去除率可达87%以上,Co2+的协同催化作用促进了Fe2+再生和HO·生成。
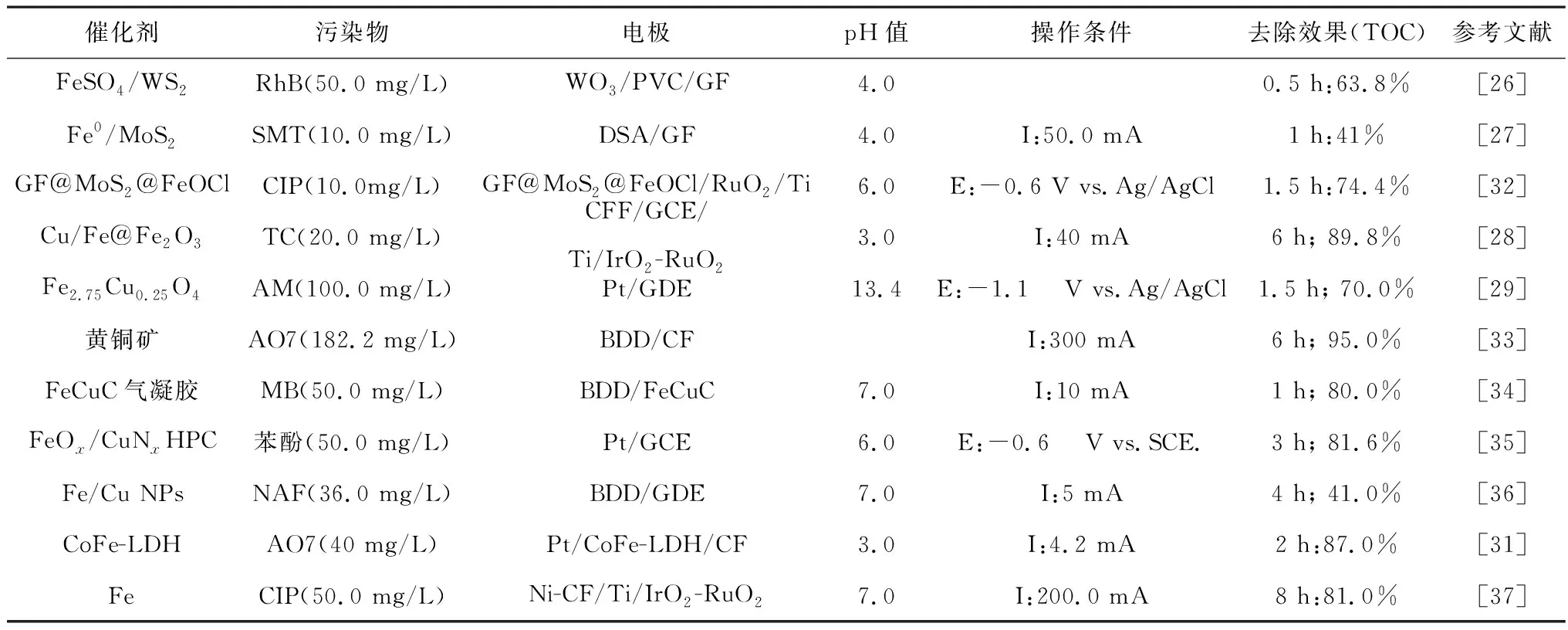
表3 Fe-M双金属催化剂电芬顿体系去除难降解有机物的效果Table 3 Effect of EF system with Fe-M bimetal catalyst in removing refractory organics
除了直接促进Fe3+还原,部分金属助催化剂通过生成其他物质来促进Fe3+/Fe2+循环。Liu等[37]制备了Ni负载CF阴极(Ni-CF)并用于强化电芬顿降解环丙沙星(CIP),Ni的引入能够明显改善TOC去除率,8 h的TOC去除率可从约42%(CF)提升至81%(Ni-CF)以上,这主要归因于Ni涂层诱导产生的大量还原性氢原子(H*)能提供电子能有效还原三价铁,促进铁循环过程,如式(5)所示。

(5)
综上,双金属协同效应对电芬顿效率的提升十分显著,但金属离子浸出以及可能造成的二次污染仍不可忽视,将过渡金属固定在载体上(常用的有碳材料)的复合催化剂能提高金属的稳定性,然而金属的负载会显著影响H2O2的生成。Cheng等[38]制备了一种OCNT封装Fe-Co-85 PBA的铠甲式催化剂,能够保证H2O2高效生成与活化,同时,碳层的保护作用进一步减少了金属离子的泄露。未来双(多)金属催化剂的研究重心仍在于研发合适的载体及其有效的复合方式,使其兼具芬顿活性位点以及两电子氧还原活性位点。此外,纳米形式的双金属催化剂具有更高的活性,但易流失、难回收的缺点限制了其在实际应用中的推广,催化剂的应用形式仍是未来的研究重点。
2.3 提高H2O2产量
O2通过2e-ORR生成过氧化氢的过程(式(1),E0=0.70 V vs.RHE)是保证电芬顿技术高效降解有机物的关键因素之一,但O2还能通过四电子氧还原反应(4e-ORR)生成H2O(式(6),E0=1.23 V vs.RHE),严重影响了H2O2的产量。施加低的应用电位有助于提高H2O2的产量,但伴随着析氢反应的出现(式(7),E0=0 V vs.RHE),电流效率进一步下降。为了提高H2O2的选择性生成,往往需要添加助催化剂引导反应向2e-ORR方向进行。

(6)
(7)
碳材料具有较大的比表面积、良好的导电性、容易修饰等优点而被广泛用作阴极,但是其H2O2选择性较差。碳材料对H2O2合成的电催化活性和选择性可以通过改变电子结构来调整[39]。Wang等[40]在碳质载体上原位合成Pd催化剂,H2O2选择性可达95%左右。除了金属负载修饰外,非金属杂原子(O、N、F等)修饰也是常见的方法[41-46]。氧分子的吸附模式对还原途径影响较大,其中末端吸附即泡林模式更容易通过2e-还原途径生成H2O2[47-48],羰基(C=O)或羧基(O=C—OH)中正电碳原子可以优先以末端吸附模式吸附氧分子,如图3所示。在碳中掺杂电负性较高的氮原子,可以通过破坏π共轭体系的完整性和诱导电荷重分布来激活碳π电子,从而改变碳材料的吸附性能,有利于H2O2的生成[45]。Su等[49]证明了引入石墨N可有效促进H2O2的生成。电负性最高的氟在碳纳米材料中掺杂后,可以诱导相邻的碳极化形成活性位点,增强O2与碳的相互作用[50]。Zhao等[51]通过控制F的含量和种类调节对氧还原反应选择性,与吸附在石墨碳上相比,中间体OOH*吸附在CF2上C的吸附能降低,有利于H2O2的生成。几种典型的碳基阴极材料及其氧还原活性位点如表4所示。
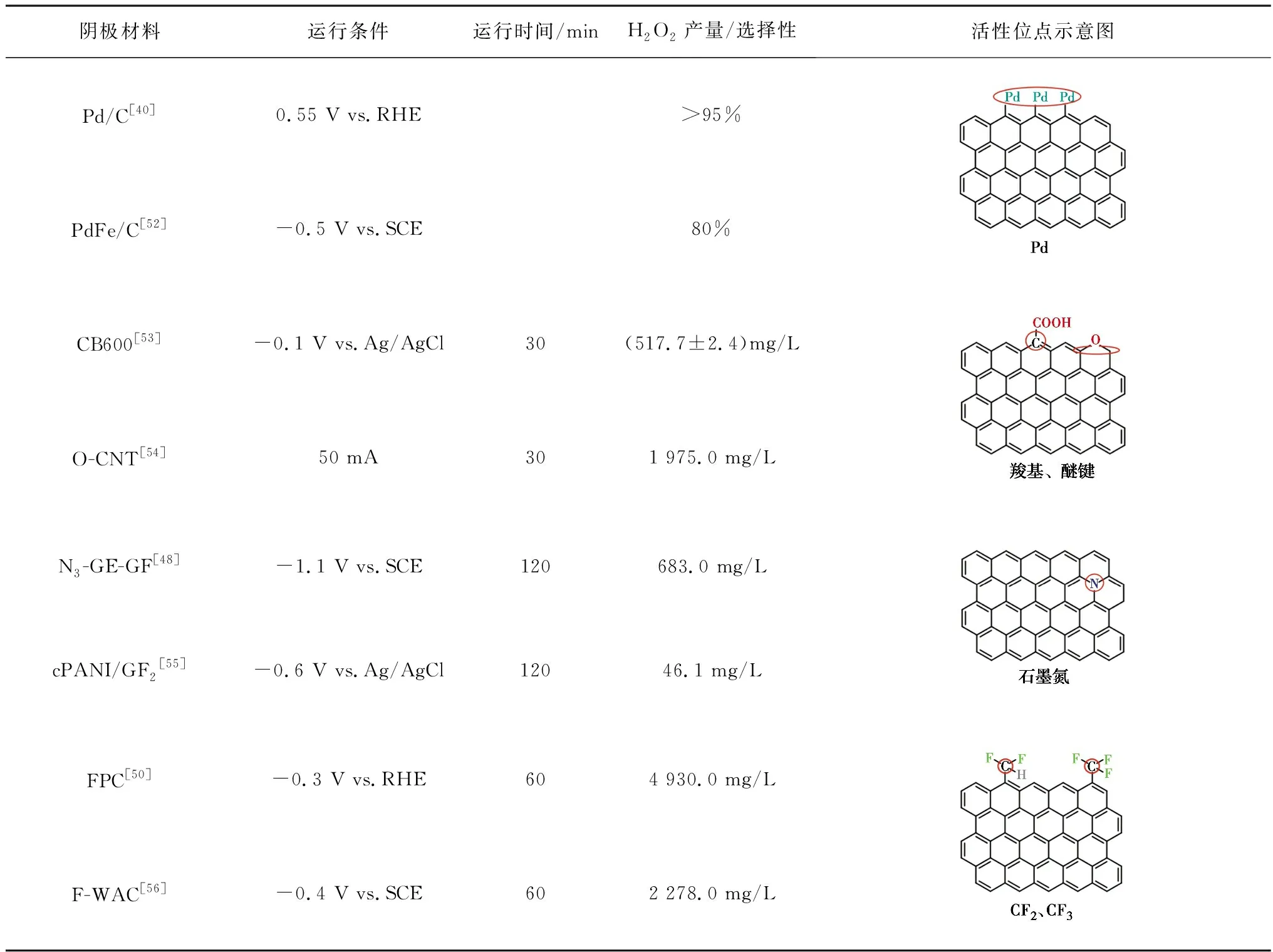
表4 典型碳基阴极材料Table 4 Typical carbon-based cathode material
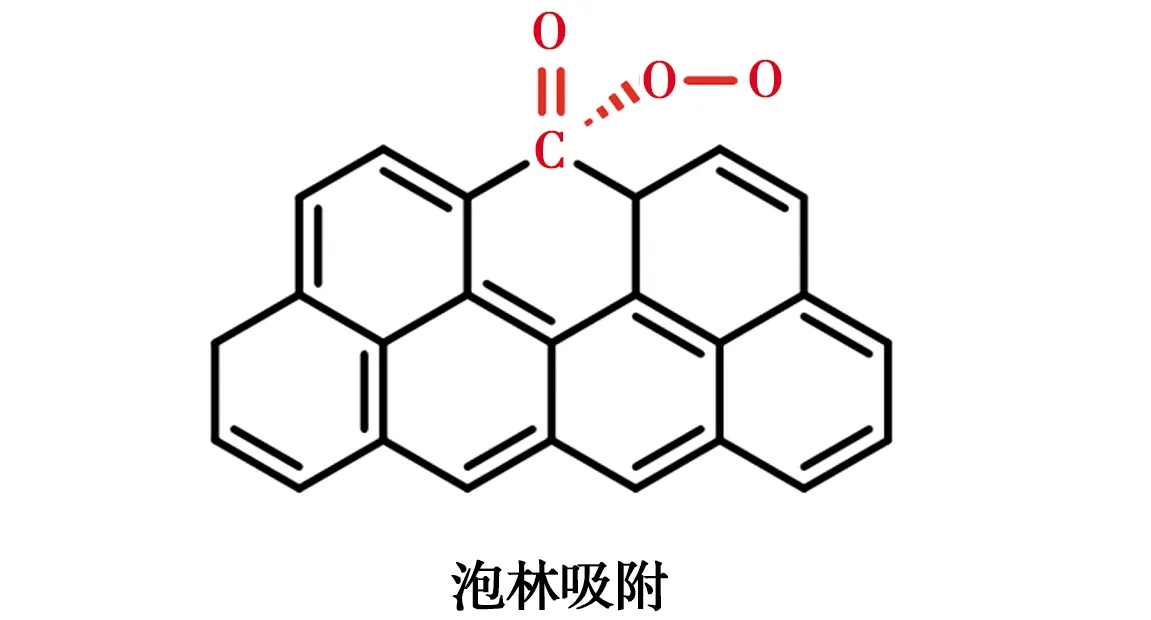
图3 双电子还原氧的泡林吸附模型Fig.3 Pauline model of oxygen adsorption toward double
各种改性碳材料被应用于电芬顿体系中。Shen等[52]将钯铁合金嵌碳气凝胶(PdFe /CA)作为阴极降解3-氯酚(3-CP),与铁碳气凝胶(Fe/CA)阴极对比发现,Pd的掺杂能够增强2e-ORR选择性,PdFe/CA通过(2+1)e-还原途径先将O2还原为H2O2,然后进一步形成HO·。PdFe /CA阴极电芬顿体系能在不添加外源性H2O2的情况下完全矿化50 mg/L的3-CP。非金属杂原子掺杂可以避免金属离子二次泄露问题,Zhang等[53]通过简单的煅烧法在炭黑表面引入含氧官能团,在-0.1 V vs.Ag/AgCl 的电位下,CB600 FAC(600 ℃煅烧得到的材料)30 min产生(517.7±2.4)mg/L的H2O2,约是未经煅烧处理CB FAC(65.3±5.6)mg/L的8倍,用于电芬顿降解50 mg/L的RhB,2 min内去除率高达91.1%。Cao等[43]在不同温度下高温碳化NH2-MIL-88B(Fe)制备了嵌入氧化铁颗粒的含氮分层多孔碳 (FeOX/NHPCt,t为碳化温度),在-0.3~-0.8 V vs.SCE电位下,FeOX/NHPC750的H2O2选择性达到95%~98%,电子转移数为2.04~2.08,是近似的2e-ORR过程。Lu等[57]以MIL-100 (Fe)∶PANI质量比为2∶1制备了Fe2O3/N-C催化剂,在溶液中检测出55 mg/L的H2O2,2 h内能完全去除10 mg/L的双酚A。MOF衍生物保留的孔结构提供了更多的活性位点以及增强传质过程,有利于高效去除污染物。
由于掺杂O、N、F阴极材料的H2O2选择性与其形态、含量密切相关,因此,如何有效调控掺杂元素的形态以及比例是未来的研究重点。与此同时,目前已有的调控手段较为复杂,成本大幅增加,开发简单的合成方法有助于提高实际应用价值。另外,强氧化性环境对催化剂稳定性的影响规律以及对催化位点的失活机制分析仍有待加强。
3 结论与展望
电芬顿技术是一项具有广阔应用前景的废水处理技术,无需投加化学试剂,对污染物去除效率高,通过添加助催化剂能改善铁离子稳定性、加速Fe3+/Fe2+循环以及提高H2O2产量,从而显著增强难降解有机污染物的去除性能。然而,运行成本高、催化剂稳定性差以及合成工艺复杂、纳米催化剂易流失、难以回收等问题仍将限制其大规模应用推广。助催化剂强化电芬顿技术的改良与发展,仍需侧重以下几个方面:
1)结合计算、表征等手段深入剖析助催化剂强化电芬顿的作用机理以及催化剂的失活机制,进一步解决因催化位点破坏而导致的催化活性降低和二次污染问题。
2)开发简单、低成本的助催化剂制备方法,并着重研发多功能型助催化剂,兼具两电子氧还原催化位点和芬顿活性位点,从而简化电芬顿工艺流程。
3)探究高效稳定的催化剂应用形式与高效稳定型反应器,减少在运行过程中催化剂的损失以及提高重用性,进而降低工程投资和运行成本。
4)联合电芬顿技术以及其他技术,充分发挥联合技术氧化还原的性能优势,实现复合污染物的协同去除。
