基于UPLC-Q-TOF-MS技术的当药化学成分分析
2021-09-06段吉平段琼2a王铁战马春艳刘永利冯丽
段吉平,段琼,2a,王铁战,马春艳,刘永利*,冯丽
1.河北省药品检验研究院,河北 石家庄 050011;
2.河北医科大学,河北 石家庄 050017
当药Swertiae Herba 为龙胆科獐牙菜属瘤毛獐牙菜Swertia pseudochinensisHara 的干燥全草[1],于夏、秋季节采挖,在河北、内蒙古药用时曾被称为“肝炎草”。《中华人民共和国药典》2020 年版中记载其味苦,性寒,归肝、胃、大肠经,用于湿热黄疸、胁痛、痢疾腹痛、食欲不振[2]。其在《内蒙古中草药》[3]、《中华本草》[4]、《全国中草药汇编》[5]、《河北植物志》(第二卷)[6]中均有记载。目前,对当药的研究相对较少,化学成分方面也鲜见报道。本实验采用超高效液相色谱-四级杆-飞行时间质谱法(UPLC-Q-TOF-MS)对当药进行化学成分分析,同时对多类别的成分进行结构鉴定及确认,为当药化学成分和药效物质基础研究及多元质量评价方法的建立提供参考。
1 材料
1.1 试药
当药药材购自安国药材市场,经河北省药品检验研究院牛小莲主任药师鉴定为龙胆科獐牙菜属瘤毛獐牙菜Swertia pseudochinensisHara的干燥全草。
对照品龙胆苦苷(批号:770-200105)、当药苷(批号:111742-200501)、獐牙菜苦苷(批号:0785-200203)、芒果苷(批号:111607-201503)、齐墩果酸(批号:110709-201808)、甜菜碱(批号:110894-200503)和槲皮素(批号:100081-201610)均购自中国食品药品检定研究院,纯度均大于99%。
甲醇(分析纯,天津科密欧化学试剂有限公司);质谱用甲酸(Fisher 公司);乙腈和甲醇(色谱纯,Merck公司)。
1.2 仪器
LC-TripleTOF®5600+型液质联用仪(SCIEX 公司),配备Analyst®TF 1.6 质谱操作软件、PeakView 2.0 质谱图分析软件;AG245 型电子天平(Mettler Toledo公司);KQ-400KDE型超声清洗仪(昆山超声仪器有限公司)。
2 方法
2.1 色谱条件
色谱柱为菲罗门Kinetex XB-C18100A(100 mm×2.1 mm,2.6 μm)。流动相为0.1% 甲酸水溶液(A)-乙腈(B),梯度洗脱(0~4 min,15%B;4~7 min,15%~30%B;7~10 min,30%~40%B;10~11 min,40%~50%B;11~30 min,50%~70%B;30~40 min,70%~100%B;40~42 min,100%B;42.1~45 min,15%B)。柱温:20 ℃,流速:0.3 mL·min-1,进样量:1 μL。
2.2 质谱条件
采用电喷雾离子源(ESI),正离子和负离子扫描模式。正离子模式下电喷雾电压为5500 V,气帘气(CUR)为30 psi(1 psi≈6.79 kPa,下同),去簇电压(DP)为80 V,碰撞能量(CE)为10 V;负离子模式下电喷雾电压为4500 V,CUR 为30 psi,DP为80 V,CE为10 V。扫描质量数为m/z100~1000。
2.3 溶液的制备
2.3.1 对照品溶液制备 取7 种对照品适量,分别加甲醇制成质量浓度为10 μg·mL-1的单一对照品溶液。
2.3.2 供试品溶液制备 取当药适量,粉碎,过三号筛,混匀,取0.5 g,精密称定,置具塞锥形瓶中,精密加入甲醇25 mL,密塞,称定质量,超声处理15 min,放冷,再称定质量,用甲醇补足减失质量,摇匀,用0.22 μm微孔滤膜滤过,即得。
2.4 化学成分分析
按2.1 和2.2 项下分析条件,对当药样品进行正、负离子模式分析,使用PeakView 2.0 质谱图分析软件对所得到的样品进行处理并对色谱峰进行同位素及MS/MS 碎片分析。首先,与对照品图谱中各已知成分的相对保留时间(tR)、准分子离子、二级质谱碎片等进行比对,再结合现有文献中同属植物的化学物质建立样品的Session 筛选库[7],对样品中的色谱峰进行解析,以满足质量误差<5×10-6、同位素分布正确且有二级碎片的化合物作为目标物质。逐一分析物质的同位素信息和MS/MS碎片碎裂规律,并结合Formula Finder 分析结果,推断可能的候选化合物[8-15]。
3 结果
3.1 色谱峰的鉴定
当药样品正、负离子模式下得到的总离子流图(TIC)见图1。共确认29 个化合物,包括7 个环烯醚萜类、13 个酮类、4 个黄酮类、2 个三萜类、2 个生物碱类和1 个核苷类成分。其中10 个为当药中未见报道的成分,见表1。
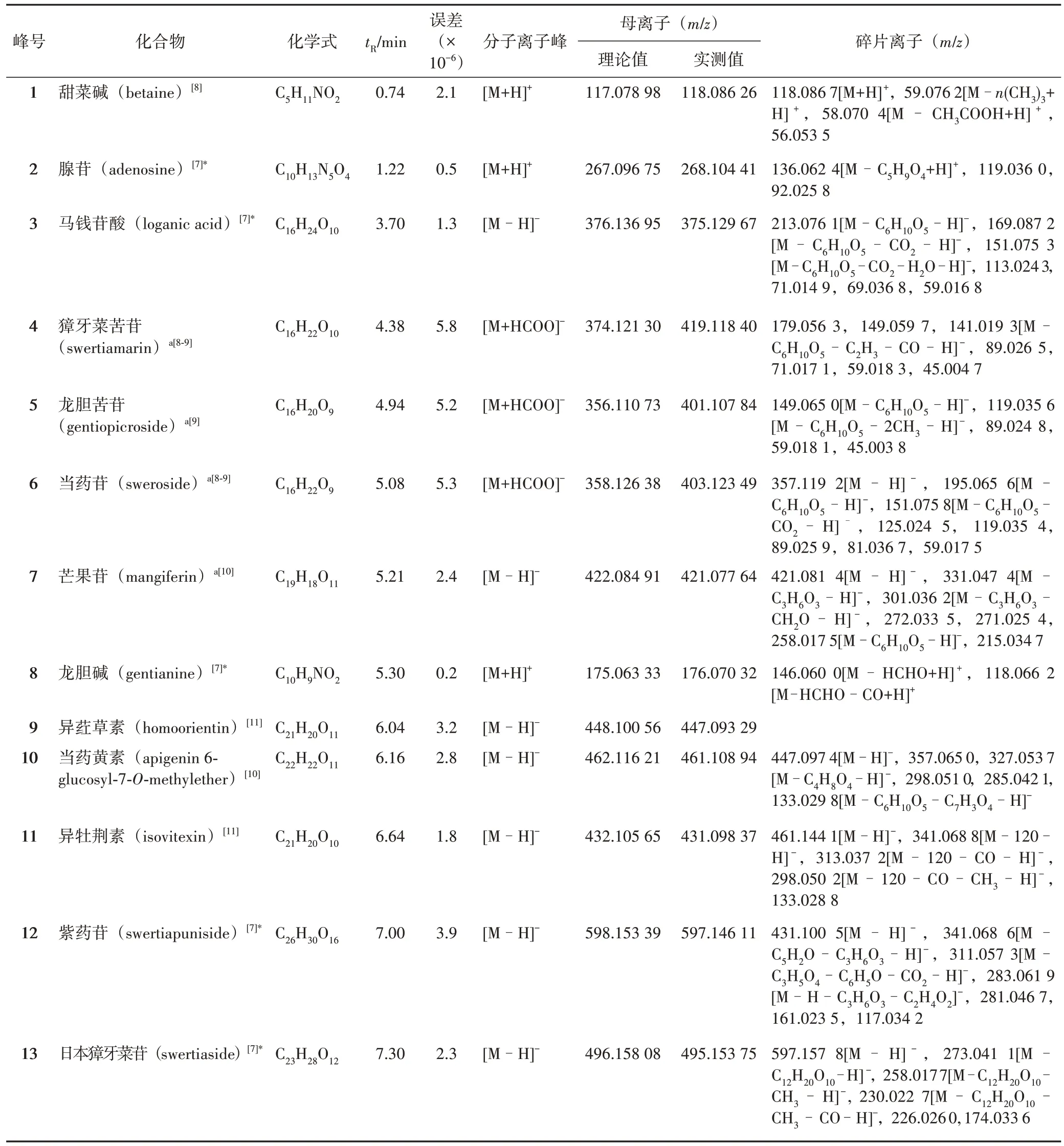
表1 当药中确认的29个成分信息

续表1
3.2 各类化合物分析鉴定
3.2.1 环烯醚萜类化合物 从当药中鉴定出的环烯醚萜类成分主要有獐牙菜苦苷、龙胆苦苷、当药苷、苦当药苷、苦龙苷、马钱苷酸和日本獐牙菜苷7 个成分。环烯醚萜类成分主要具有特征的二氢吡喃环顺式连接1 个五元环的单元结构,而当药中主要存在环戊烷型和裂环烯醚萜苷2 种类型。在所采用的色谱系统中加入了甲酸,故环烯醚萜类物质准分子离子峰存在[M+HCOO]-及[M-H]-2 种形式。当药中的环烯醚萜类结构均存在-C6H10O5碎片丢失,同时因裂环烯醚萜苷类化合物具有半缩醛结构,化学性质活泼,在糖苷键断裂后继而发生逆狄尔斯-阿德尔(RDA)裂解,失去-CO2、-H2O 等碎片,同时可能发生-CH3的丢失。以当药苷为例,m/z403.123 5[M+HCOO]-与m/z357.119 2[M-H]-同时存在于系统中,但[M+HCOO]-响应明显强于[M-H]-。在丢失-C6H10O5碎片后,继而发生RDA 裂解,脱去-CO2,得到m/z151.075 8[M-C6H10O5-CO2-H]-。
3 号色谱峰保留时间为3.70 min,根据PeakView 2.0 质谱图软件分析化学式为C16H24O10,理论质量数为376.136 95[M],在负离子检测模式下实际测得质量数为m/z375.131 63[M-H]-。其进一步脱葡萄糖基产生m/z213.076 1[M-C6H10O5-H]-,由于羧基的不稳定性,[M-C6H10O5-H]-脱去羧基得到m/z169.087 0[M-C6H10O5-CO2-H]-,再脱去中性小分子H2O,得到m/z151.075 3 [C9H11O2]-。该化合物与文献报道的马钱苷酸碎片信息一致[17],推测其为马钱苷酸。从其MS/MS 图中所得到的碎片离子信息可解析其裂解方式,见图2。

图2 马钱苷酸裂解途径
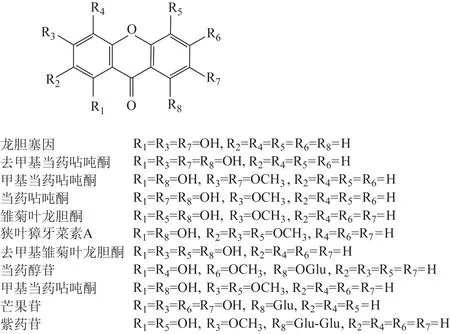
图3 当药中的11个单酮及酮苷
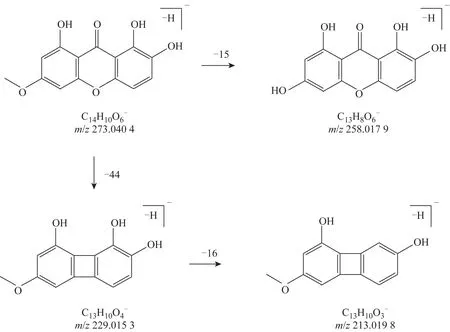
图4 当药呫吨酮裂解途径
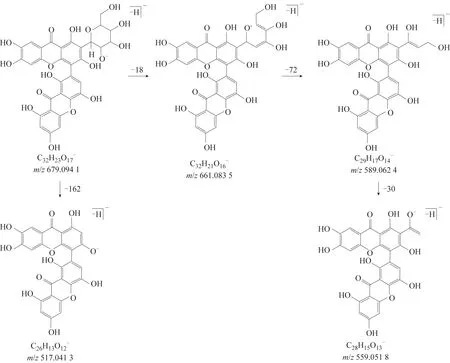
图5 3-O-去甲基紫药双呫吨酮苷裂解途径
具有O苷键的酮苷中,因糖苷键的不稳定性,首先生成带1个负电荷的酮苷元。12号色谱峰保留时间为7.00 min,根据PeakView 2.0质谱图软件分析化学式为C26H30O16,理论质量数为598.153 39[M],在负离子检测模式下实际测得为m/z597.146 11[M-H]-。母离子脱去-C12H20O10,生成的碎片离子为m/z273.041 1[M-C12H20O10-H]-。m/z273.041 1[M-C12H20O10-H]-在丢失-CH3碎片后,继而丢失-CO,产生m/z258.017 7[M-C12H20O10-CH3-H]-及m/z230.022 7 [MC12H20O10-CH3-CO-H]-的碎片。推测该化合物可能为紫药双呫吨酮苷。
3.2.3 黄酮类化合物 当药中主要鉴定出4 个黄酮类化合物,包括当药黄素、异牡荆素、异荭草素和槲皮素。在负离子扫描条件下,黄酮类成分易中性丢失-CO、-CO2,并可能在糖基的部分发生RDA裂解。10号色谱峰保留时间为6.16 min,根据PeakView 2.0质谱图软件分析化学式为C22H22O11,理论质量数为462.116 21[M],在负离子检测模式下实际测得质量数为m/z461.108 94[M-H]-。母离子发生RDA反应,生成了m/z341.068 8[M-120-H]-的碎片,随后又丢失-CO及-CH3,得到m/z313.037 2[M-120-CO-H]-和m/z298.050 2[M-120-CO-CH3-H]-。推测该化合物为当药黄素。
3.2.4 三萜类化合物 28 号色谱峰保留时间为27.60 min,根据PeakView 2.0 质谱图软件分析化学式为C30H48O3,其一级和二级质谱图与齐墩果酸对照品一致。齐墩果酸理论质量数为m/z456.360 3[M],在负离子检测模式下实际测得质量数为m/z455.353 07[M-H]-,母离子经过脱羧反应、失去活泼氢和1 分子H2O,形成了共轭结构的稳定碎片离子m/z407.334 4[M-H-HCOOH-2H]-和m/z391.304 3[MHCOOH-H2O-H]-碎片离子。
29 号色谱峰保留时间为29.50 min,根据PeakView 2.0 质谱图软件分析化学式为C30H48O,理论质量数为424.370 52[M]。在正离子检测模式下测得质量数为m/z425.377 79[M+H]+。母离子发生RDA裂解,生成m/z301.258 3[M-124]+的碎片,推测其结构为蒲公英赛-14-烯-3-酮。
3.2.5 生物碱类化合物 1 号色谱峰保留时间为0.74 min,根据PeakView 2.0质谱图软件分析化学式为C5H11NO2,其保留时间、一级和二级质谱图与甜菜碱对照品一致。甜菜碱理论质量数为m/z117.078 98[M],在正离子模式下,实际测得质量数为m/z118.086 26[M+H]+,母离子发生化学键断裂及重排失去m/z59.073 5[N(CH3)3]+和m/z60.021 1 [CH3COOH]+的碎片,得到m/z59.076 2[M-N(CH3)3+H]+和m/z58.070 4[M-CH3COOH+H]+的碎片离子。
8 号色谱峰保留时间为5.30 min,根据PeakView 2.0质谱图软件分析化学式为C10H9NO2,理论质量数为175.063 33[M],在正离子模式下,实际测得质量数为m/z176.070 61[M+H]+。母离子发生化学键断裂及重排失去HCHO,得到m/z146.060 0[MHCHO+H]+,随后[M-HCHO+H]+丢失CO,得到m/z118.066 2 的碎片离子,推测结构为龙胆碱,裂解途径见图6。
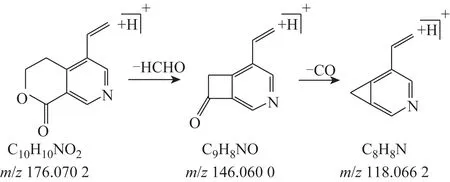
图6 龙胆碱的裂解途径
3.2.6 核苷类化合物 29 号色谱峰保留时间为1.22 min,根据PeakView 2.0 质谱图软件分析化学式为C10H13N5O4,理论质量数为267.096 75[M],在正离子模式下,实际测得质量数为m/z268.104 41[M+H]+,母离子发生化学键断裂及重排失去C5H9O4,得到m/z136.062 4的碎片离子。推测结构为腺苷。
4 讨论
本研究应用UPLC-Q-TOF-MS、Analyst®TF 1.6 Software 质谱操作软件和PeakView 2.0 质谱图分析软件对当药的化学成分进行分析,建立了獐牙菜属植物的物质筛选库,对样品中色谱峰进行解析,分析物质的同位素信息和MS/MS 碎片信息,与对照品图谱中各已知成分的保留时间、准分子离子、二级质谱碎片等进行比对,再结合现有文献中同属植物的化学物质[7],对样品中的色谱峰进行解析。最终确认了当药中的7个环烯醚萜类、13个酮类、4个黄酮类、2 个三萜类、2 个生物碱类和1 个核苷类成分。其中马钱苷酸、龙胆碱、紫药双呫吨酮苷、日本獐牙菜苷、3-O-去甲基紫药双呫吨酮苷、槲皮素、龙胆赛因、狭叶獐牙菜素A、腺苷和蒲公英赛-14-烯-3-酮为首次在当药中报道。
